
Peptide length significantly influences in vitro affinity for MHC class II molecules
- Research
- Open Access
Peptide length significantly influences in vitro affinity for MHC class II molecules
- Cathal O'Brien1_26,
- Darren R Flower2_26 and
- Conleth Feighery1_26Email author
- Received: 26 June 2008
- Accepted: 26 November 2008
- Published: 26 November 2008
Abstract
Background
Class II Major Histocompatibility Complex (MHC) molecules have an open-ended binding groove which can accommodate peptides of varying lengths. Several studies have demonstrated that peptide flanking residues (PFRs) which lie outside the core binding groove can influence peptide binding and T cell recognition. By using data from the AntiJen database we were able to characterise systematically the influence of PFRs on peptide affinity for MHC class II molecules.
Results
By analysing 1279 peptide elongation events covering 19 distinct HLA alleles it was observed that, in general, peptide elongation resulted in increased MHC class II molecule affinity. It was also possible to determine an optimal peptide length for MHC class II affinity of approximately 18–20 amino acids; elongation of peptides beyond this length resulted in a null or negative effect on affinity.
Conclusion
The observed relationship between peptide length and MHC class II affinity has significant implications for the design of vaccines and the study of the epitopic basis of immunological disease.
Keywords
- Major Histocompatibility Complex
- Major Histocompatibility Complex Class
- Major Histocompatibility Complex Molecule
- Binding Groove
- Major Histocompatibility Complex Allele
Background
Classical major histocompatibility complex (MHC) molecules are divided into two groups: class I and class II [1]. Each contains large numbers of alleles, all of which bind antigen for cell surface presentation to T cells. MHC class I molecules comprise a single polymorphic chain coupled to a single conserved protein: β2 microglobulin. In contrast, MHC class II molecules comprise two polymorphic subunits: an α and a β chain. A notable difference between the classes is the open-ended peptide-binding groove of MHC class II molecules compared to the closed binding site of MHC class I. This feature, which has been observed in many X-ray crystallographic structures, has been used to explain the observed differences in peptide length accommodated by the two classes [2, 3, 4]. Class I molecules typically accommodate peptides of eight to ten residues, although instances of longer peptide binding have now been reported [5].
In contrast, MHC class II molecules can accommodate much longer peptides. Stern et al. investigated an influenza-derived peptide complexed with HLA-DR1; they saw the peptide bound in an extended conformation with five binding pockets, which engaged peptide side chains, while the flanking regions extended out of the groove [3]. Since MHC class II molecules have an open-ended binding groove, they do not, in general, restrict the length of bound peptides. The length of peptides occupying the groove can vary considerably as a consequence of semi-stochastic proteolytic degradation [6].
Several studies have shown the influence of residues outside the main nonameric core on binding and subsequent T cell recognition. Residues outside the nonameric binding region but within an extended binding groove have been called peptide-flanking residues (PFRs) [7]. The importance of PFRs for T cell recognition has also been investigated. It is widely accepted that these flanking regions can contribute to T cell mediated pMHC recognition [8]. Arnold and colleagues showed that certain T cell responses were completely dependent on residues at peptide positions P-1 and P11. Similar findings were reported by Stepniak et al for immunostimulatory HLA-DQ2 binding peptides derived from gliadin and glutenin proteins [9]. The findings of both authors can be partly explained by the results of X-ray crystallographic studies of peptide-MHC class II-TCR trimolecular complexes [10, 11]. These reports indicate that the CDR3 regions of the TCR α and β chains tend to locate over the p5 residue in the binding groove allowing the TCR to interact with the PFRs. It has also been shown that the properties of residues in the PFRs can influence T cell recognition, with residues capable of forming salt-bridges or hydrogen bonding with the TCR being most favoured [8].
Much circumstantial if anecdotal evidence suggests that PFRs contribute strongly to peptide-MHC stability. They can provide an increased measurable affinity of peptide for the MHC binding groove, in particular the residue at position P-1 [12]. Nelson et al. reported that the effect of increased stability was greater for a peptide length increase of six amino acids compared to four. They also found the effect to differ depending on which terminus was elongated. Moreover, a previous study by Srinivasan et al showed increased affinity resulting from the peptide elongation of a single peptide [13]. How such peptide elongations might increase affinity remains unclear. Although it is theoretically possible PFRs interact with the MHC molecules outside the groove, this has yet to be shown experimentally, nor has it been seen in X-ray crystallographic structures.
We have sought to elucidate this phenomenon further, by examining the relationship between peptide length and MHC class II affinity. We devised custom algorithms and applied them to data available in the AntiJen database [14] to firstly ascertain whether peptide length can impact on affinity of a peptide for an MHC class II molecule. This was assessed by searching the database for instances of peptide elongation and determining the reported effect on MHC class II affinity. Following this, we examined whether there was a demonstrable limit to the effect of peptide length on MHC class II affinity and subsequently determined whether any effects on affinity were peptide terminus specific. Our analysis indicated the positive impact of peptide length on affinity. We also identified an optimal length for peptide-MHC class II binding and demonstrated that the effects on peptide affinity are terminus independent.
Results
Most peptide elongation events result in enhanced affinity
Chi Square Table for the observed elongation events.
Amino |
Both |
Carboxy |
All |
|
|---|---|---|---|---|
Decreased (Observed) |
120 |
80 |
92 |
292 |
Decreased (Expected) |
120.3 |
81.5 |
90.2 |
292 |
Increased (Observed) |
407 |
277 |
303 |
987 |
Increased (Expected) |
406.7 |
275.5 |
304.8 |
987 |
All |
527 |
357 |
395 |
1279 |
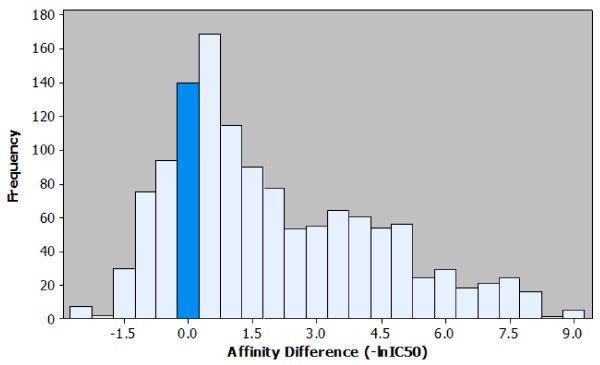
Histogram of the affinity differences observed in the peptide elongation dataset. Observed effects on affinity for each peptide elongation are plotted based on the frequency of occurrence. It is possible to visualise the most common affinity difference (+0.5), the most extreme affinity difference (+9.0) and the ratio of positive to negative effects. For ease of interpretation the bar representing the number of instances where no effect on affinity was observed is highlighted.
Peptide elongation below an optimal length results in enhanced affinity
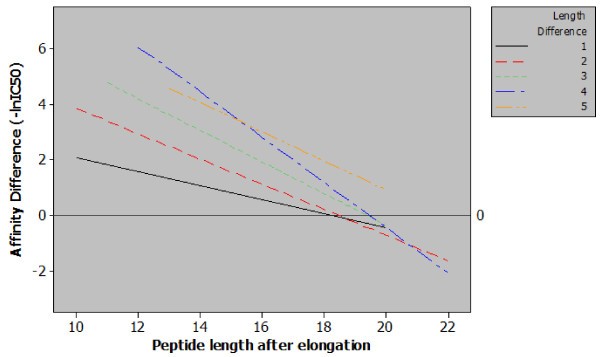
Regression plot of classes of peptide length additions. Five regression lines relating observed differences in affinity to final peptide length for each magnitude of peptide elongation. The content of this figure shows two important points relating sequence length and MHC class II affinity. Firstly, when correcting for final peptide length a greater length addition will generate a greater increase in affinity e.g. for peptides with a final length of 14 amino acids increasing the length by four residues has a much greater effect than a single residue increase. Secondly, from the included data it is possible to observe a length of between 18 and 20 amino acids, beyond which, increasing the length of the peptide has a null or negative effect.
The observed effect is terminus independent
Composition of the dataset of elongation events.
Allele |
Count |
Percent |
|---|---|---|
DPB1*0401 |
38 |
2.97 |
DPB1*0402 |
38 |
2.97 |
DQA1*0301/DQB1*0301 |
64 |
5 |
DQA1*0501/DQB1*0201 |
114 |
8.91 |
DRB1*0101 |
124 |
9.7 |
DRB1*0301 |
1 |
0.08 |
DRB1*0401 |
77 |
6.02 |
DRB1*0404 |
2 |
0.16 |
DRB1*0405 |
3 |
0.23 |
DRB1*0802 |
3 |
0.23 |
DRB1*0803 |
2 |
0.16 |
DRB1*0901 |
3 |
0.23 |
DRB1*1101 |
3 |
0.23 |
DRB1*1201 |
3 |
0.23 |
DRB1*1302 |
3 |
0.23 |
DRB1*1501 |
682 |
53.32 |
DRB3*0101 |
24 |
1.88 |
DRB4*0101 |
1 |
0.08 |
DRB5*0101 |
94 |
7.35 |
Total |
N = 1279 |
100 |
Discussion
While MHC class II molecules and their ligands are well characterised, the impact of peptide length on MHC affinity is not well understood. While several anecdotal observations have purported to show that peptide length may influence affinity we have for the first time systematically quantified this relationship using in silico analysis. Using novel computational analyses, we exploit the wealth of manually-curated quantitative binding data available within the AntiJen database [14].
When attempting to derive a relationship between sequence length and observed MHC affinity, one is limited by several factors. One such factor is the presence of different binding determinants in most sequences that could significantly bias any simple length-affinity correlation. Thus, if one examines the affinity of two sequences of different length and low sequence similarity, one cannot ascertain if an altered affinity is due to differences in the binding determinant or to changes in sequence length. To isolate such a relationship, one must normalise all factors (such as MHC allele, experimental conditions, and peptide sequence) except for sequence length before analysing the affect on affinity. To that end, we identified examples of affinity measurements, for a single sequence and an elongated equivalent, which were performed under the same experimental conditions. The benefits of this approach were twofold. Firstly, inter-laboratory variation in affinity measurements would be excluded. Secondly, bias from variations in the binding determinant would also be reduced if not eliminated, thus removing bias from intra- but not inter-sequence comparisons. Bias from inter-sequence comparisons is unavoidable. We optimised for the required task in order to avoid representing bias as effect.
The data presented in Figure 2 allowed us to examine the limits of the effect with respect to final peptide length. By examining the log fold-change in affinity for MHC class II as the length of the peptide increases it can clearly be seen that the impact on affinity lessens. The regression plot also shows that at approximately 19 amino acids the effect of peptide elongation on affinity becomes zero. Elongating peptides beyond this point would – in the majority of cases – result in a diminished affinity. This seems logical: if no such ceiling existed for the positive effect of peptide length then sequence length alone would be the key determinant of peptide-MHC affinity. This discovery is also strengthened by the findings of Lippolis et al. who eluted and sequenced peptides from HLA-DR*0401 and found that the most abundant species within a family of nested peptides were between 14 and 21 residues in length [15]. Our findings do not however suggest that a single optimal length would exist for all peptides for all MHC class II alleles, and indeed the relative heterogeneity of peptides binding to MHC class II molecules is well reported [15, 16, 17]. However, this approximation to an optimal length is consistent for a large number of peptide sequences and a large number of MHC class II alleles.
Many explanations for this length ceiling present themselves. It may be that as peptides become very long they are no longer able to form favourable interactions with the MHC or TCR. Alternatively, when MHC class II binding peptides attain a certain length they may form secondary or tertiary structures outside the binding site that are less compatible with MHC class II binding. An exception to this rule may be the 33-amino acid peptide initially described by Shan et al [18]. It binds to HLA-DQ2 and stimulates disease associated T cells. Despite its unusual length, this peptide retains its MHC class II binding abilities as its proline rich nature helps it to form a type II polyproline helix. This is the conformation normally seen in MHC class II binding grooves [3].
Our results clearly imply a general correlation between increasing MHC binding affinity and increasing peptide length. It is not peculiar to a restricted subset of peptides. Sercarz and Maverakis posited that longer peptides have a greater binding affinity for MHC class II [19]; this assertion was based on observations from a few articles, e.g. the study by Srinivasan et al [13]. However, to the best of our knowledge, ours is the first study to test this hypothesis systematically for several MHC class II alleles. The results of our study are compelling: across 1279 identified peptide elongation events for peptides binding to 19 distinctive MHC class II alleles, affinity predominately increased. It is important to note, however, that the examined peptides are more representative of the HLA-DR species than either HLA-DP or HLA-DQ, and this may result from an apparent publication bias in favour of HLA-DR which will be reflected in the AntiJen database.
Our findings have clear implications for experimental immunology and vaccinology, as well as computational immunology; and these implications are many. Increased affinity in vivo may result in increased reactivity to recently degraded peptides. Presumably, recently degraded peptides may be less completely digested and – as a result of increased length – have an increased affinity for MHC class II relative to shorter more fully degraded peptides. Such an effect would, in theory, increase the likelihood of T cells recognising more recently endocytosed peptides rather than a background of self-peptides. Additionally, the ability of exopeptidases to digest termini of peptides within the MHC class II binding cleft [6] may effectively reduce their MHC class II affinity. Thus, peptides susceptible to digestion by exopeptidases may prove to be less immunogenic in vitro. The field of subunit vaccine design may be able to augment vaccine efficacy by increasing peptide length or eliminating potential proteolytic cleavage sites, which may result in an inability of normal proteolytic enzymes to digest peptides to shorter equivalents with reduced immunogenicity.
Conclusion
Using an in silico approach we were able to define a relationship between peptide length and MHC class II affinity. The results of this study demonstrate that, within limits, elongation of a peptide is likely to result in a greater affinity for MHC class II molecules. It was also possible to show that the optimal length for peptide-MHC affinity was approximately 18–20 amino acids. Additionally, it was also possible to demonstrate that the effects are consistent regardless of which terminus was elongated. Given the well-characterised importance of peptide-MHC interaction to the cellular immune response, it is likely that the findings of this study will prove of significance in vaccine design and the epitopic basis of immunological disease.
Methods
Dataset formation
Data for the study were obtained by querying the AntiJen database [14] for all sequences known to bind human MHC class II molecules. These data were imported into a spreadsheet and then further filtered to exclude those data for which no radiometrically determined IC50 value were present. IC50 values were then transformed to their -lnIC50. This dataset was then output to a text file containing epitope sequence, -lnIC50 value, pubmed ID of the source article, experimental conditions and the MHC binding allele. This database formed the source data for interrogation using a bespoke algorithm.
Data query algorithm
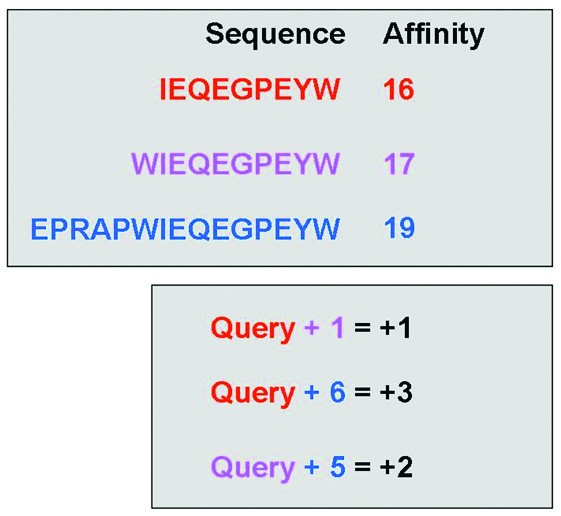
Illustration of the elongation event search algorithm. The upper section contains theoretical affinity data for a set of peptides within a single publication. The lower section shows the differences in peptide length and associated changes in affinity (colour coded by peptide). The algorithm searches within a publication for any instance of a sequence exhibiting a longer counterpart. The algorithm then returns the length difference and affinity difference for each recorded elongation event (i.e. Query+1 = +1, and Query +6 = +3).
Statistical analysis of elongation data
All statistical analyses were performed using the Minitab® software package (Minitab Inc) for statistical analysis. Discrete datasets were isolated for particular analyses such as the chi-square analysis using Microsoft Excel®.
Declarations
Acknowledgements
Funding for this study was provided by the Department of Immunology, Trinity College Dublin.
Authors’ Affiliations
References
- Janeway C: Immunobiology 5 Edition Garland Scientific 2001.Google Scholar
- Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC: Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368:711–718.View ArticlePubMedGoogle Scholar
- Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC: Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 1994, 368:215–221.View ArticlePubMedGoogle Scholar
- Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC: X-ray crystal structure of HLA-DR4 (DRA *0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity 1997, 7:473–481.View ArticlePubMedGoogle Scholar
- Miles JJ, Elhassen D, Borg NA, Silins SL, Tynan FE, Burrows JM, Purcell AW, Kjer-Nielsen L, Rossjohn J, Burrows SR, McCluskey J: CTL recognition of a bulged viral peptide involves biased TCR selection. J Immunol 2005, 175:3826–3834.PubMedGoogle Scholar
- Larsen SL, Pedersen LO, Buus S, Stryhn A: T cell responses affected by aminopeptidase N (CD13)-mediated trimming of major histocompatibility complex class II-bound peptides. J Exp Med 1996, 184:183–189.View ArticlePubMedGoogle Scholar
- Moudgil KD, Sercarz EE, Grewal IS: Modulation of the immunogenicity of antigenic determinants by their flanking residues. Immunol Today 1998, 19:217–220.View ArticlePubMedGoogle Scholar
- Arnold PY, La Gruta NL, Miller T, Vignali KM, Adams PS, Woodland DL, Vignali DA: The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J Immunol 2002, 169:739–749.PubMedGoogle Scholar
- Stepniak D, Vader LW, Kooy Y, van Veelen PA, Moustakas A, Papandreou NA, Eliopoulos E, Drijfhout JW, Papadopoulos GK, Koning F: T-cell recognition of HLA-DQ2-bound gluten peptides can be influenced by an N-terminal proline at p-1. Immunogenetics 2005, 57:8–15.View ArticlePubMedGoogle Scholar
- Hennecke J, Carfi A, Wiley DC: Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. Embo J 2000, 19:5611–5624.View ArticlePubMedGoogle Scholar
- Hennecke J, Wiley DC: Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J Exp Med 2002, 195:571–581.View ArticlePubMedGoogle Scholar
- Nelson CA, Petzold SJ, Unanue ER: Identification of two distinct properties of class II major histocompatibility complex-associated peptides. Proc Natl Acad Sci USA 1993, 90:1227–1231.View ArticlePubMedGoogle Scholar
- Srinivasan M, Domanico SZ, Kaumaya PT, Pierce SK: Peptides of 23 residues or greater are required to stimulate a high affinity class II-restricted T cell response. Eur J Immunol 1993, 23:1011–1016.View ArticlePubMedGoogle Scholar
- Toseland CP, Clayton DJ, McSparron H, Hemsley SL, Blythe MJ, Paine K, Doytchinova IA, Guan P, Hattotuwagama CK, Flower DR: AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome Res 2005, 1:4.View ArticlePubMedGoogle Scholar
- Lippolis JD, White FM, Marto JA, Luckey CJ, Bullock TN, Shabanowitz J, Hunt DF, Engelhard VH: Analysis of MHC class II antigen processing by quantitation of peptides that constitute nested sets. J Immunol 2002, 169:5089–5097.PubMedGoogle Scholar
- Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DA, Strominger JL: Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature 1992, 358:764–768.View ArticlePubMedGoogle Scholar
- Hayden JB, McCormack AL, Yates JR 3rd, Davey MP: Analysis of naturally processed peptides eluted from HLA DRB1*0402 and *0404. J Neurosci Res 1996, 45:795–802.View ArticlePubMedGoogle Scholar
- Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C: Structural basis for gluten intolerance in celiac sprue. Science 2002, 297:2275–2279.View ArticlePubMedGoogle Scholar
- Sercarz EE, Maverakis E: Mhc-guided processing: binding of large antigen fragments. Nat Rev Immunol 2003, 3:621–629.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.