

Modeling the bound conformation of Pemphigus Vulgaris-associated peptides to MHC Class II DR and DQ Alleles
- Research
- Open Access
Modeling the bound conformation of Pemphigus Vulgaris-associated peptides to MHC Class II DR and DQ Alleles
- Received: 20 October 2005
- Accepted: 21 January 2006
- Published: 21 January 2006
Abstract
Background
Pemphigus vulgaris (PV) is a severe autoimmune blistering disorder characterized by the presence of pathogenic autoantibodies directed against desmoglein-3 (Dsg3), involving specific DR4 and DR6 alleles in Caucasians and DQ5 allele in Asians. The development of sequence-based predictive algorithms to identify potential Dsg3 epitopes has encountered limited success due to the paucity of PV-associated allele-specific peptides as training data.
Results
In this work we constructed atomic models of ten PV associated, non-associated and protective alleles. Nine previously identified stimulatory Dsg3 peptides, Dsg3 96–112, Dsg3 191–205, Dsg3 206–220, Dsg3 252–266, Dsg3 342–356, Dsg3 380–394, Dsg3 763–777, Dsg3 810–824 and Dsg3 963–977, were docked into the binding groove of each model to analyze the structural aspects of allele-specific binding.
Conclusion
Our docking simulations are entirely consistent with functional data obtained from in vitro competitive binding assays and T cell proliferation studies in DR4 and DR6 PV patients. Our findings ascertain that DRB1*0402 plays a crucial role in the selection of specific self-peptides in DR4 PV. DRB1*0402 and DQB1*0503 do not necessarily share the same core residues, indicating that both alleles may have different binding specificities. In addition, our results lend credence to the hypothesis that the alleles DQB1*0201 and *0202 play a protective role by binding Dsg3 peptides with greater affinity than the susceptible alleles, allowing for efficient deletion of autoreactive T cells.
Keywords
- Major Histocompatibility Complex
- Human Leukocyte Antigen
- Major Histocompatibility Complex Class
- Human Leukocyte Antigen Class
- Major Histocompatibility Complex Molecule
Introduction
Major histocompatibility complex (MHC) class II molecules are heterodimeric glycoproteins consisting of α and β chains, with approximate molecular mass of 33 kDa and 28 kDa respectively. MHC class II molecules are specialized peptide receptors that play a critical role in initiating and regulating immune responses by binding peptide fragments that are 10–30 amino acids long [1] and present them on the surface of antigen-presenting cells for recognition by CD4+ T cells. The class II region encodes genes for the human leukocyte antigen (HLA) or histocompatibility molecules class II structural genes DP, DQ and DR [2, 3]. While specific DP, DQ or DR alleles at the HLA class II locus have been shown to correlate with particular autoimmune diseases, a variety of confounding factors including strong linkage disequilibrium between the different HLA alleles, especially DR and DQ, complicates the exact identification of MHC susceptibility alleles.
Pemphigus Vulgaris (PV) is a potentially life-threatening form of autoimmune blistering skin disorder due to loss of integrity of normal intercellular attachments within the epidermis and mucosal epithelium. Like other autoimmune skin conditions such as how i cured my lichen sclerosus, PV involves immune system dysfunction. The disease is characterized by the presence of pathogenic autoantibodies directed mainly against a 130-kDa transmembrane glycoprotein, desmoglein-3 (Dsg3) [4], within the desmosomes of the spinous layer of the skin. Strong association of PV to the major histocompatibility complex class II serotypes DR4 and DR6 have been reported in the literature [5, 6, 7] with over 95% of PV patients possessing one or both of these alleles [7]. Direct nucleotide sequence analysis of DR4 and DR6 subtypes revealed that susceptibility to PV is strongly linked to DRB1*0402 and DQB1*0503 molecular subtypes, respectively [7, 8].
The use of computational techniques has been instrumental in advancing epitope-based vaccine research, with much work focusing on predicting the binding specificities of peptides to MHC molecules. Sequence-based predictive systems, based on identifying patterns in peptides with experimentally determined binding strength, are widely used to facilitate the identification of binding peptides to MHC class II molecules. Southwood et al. [9] developed a scoring matrix for DRB1*0401 based on a polynomial technique. Mallios [10, 11] reported the results of an iterative stepwise discriminant analysis meta-algorithm to identify binders from non-binders for DRB1*0101. Brusic et al. [12] applied a genetic algorithm to discriminate binders from non-binders for DRB1*0401. Noguchi et al. [13, 14] utilized both fuzzy neural network and hidden Markov model to predict potential binders to DRB1*0401 and DRB1*0101. Hammer et al. [15] employed a peptide side chain scanning technique for screening peptides that interact with DRB1*0401. Nielsen et al. [16] used a Gibbs sampling method for discriminating DRB1*0401 specific binders from non-binders. Karpenko et al. [17] made use of an ant colony system to search for DRB1*0401 binding and non-binding peptides. Doytchinova and Flower [18] employed an additive method for predicting the binding affinity of peptides bound to DRB1*0401, DRB1*0101 and DRB1*0701 based on the sum of the contributions of the amino acids at each position of the bound peptide and various interactions between them. However, despite recent advances in sequence-based predictive techniques, computational models for the majority of PV implicated alleles have been lacking, mainly due to the paucity of sufficient peptides as training data, and are unsuitable for predicting peptide binding to PV implicated alleles. Also, most computational methods focus on predicting just peptide binders and non-binders, whereas our aim is to distinguish between different modes of binding conferred by susceptible and protective alleles.
An alternative approach to predicting peptide/MHC (pMHC) complexes without the need of a large training dataset is to use information derived from three-dimensional structures. Logean and Rognan [19] utilized a combinatorial built-up algorithm to construct the three-dimensional structure of pMHC complexes. Altuvia et al. [20] reported the use of a computational threading approach to rank potentially binding peptides to MHC class I molecules. Lim et al. [21] employed molecular dynamic simulations to examine the structures of A*0201 in complex with 9-mer peptides. Michielin et al. [22, 23] applied homology modeling to select peptides that bind to A*0201.
In addition to predicting the binding specificities of peptides to MHC molecules, three-dimensional models have also been used for structural classification of alleles into HLA "supertypes" based on structural features derived from the binding sites. Recently, Doytchinova et al. [24, 25] employed hierarchical clustering and principal component analysis to classify alleles based on structural features into eight HLA class I and twelve HLA class II supertypes. The structural classification of alleles facilitates the identification of allelic subgroups that may share similar binding specificities and shed light into their possible role in cellular immunity against pathogens.
In the present study, we have attempted to understand the functional correlation between MHC class II alleles and PV, from a structural interaction view point. Molecular modeling of ten PV associated and non-associated MHC class II receptors (DR4: DRB1*0401, *0402, *0404, *0406, DR6 (also classified now as DR14): DRB1*1401, *1404, *1405, DQ2: DQB1*0201, *0202 and DQ5: DQB1*0503) were performed to explore the structural organization of the binding groove of these alleles. Nine previously identified epitopes, Dsg3 96–112, Dsg3 191–205, Dsg3 206–220, Dsg3 252–266, Dsg3 342–356, Dsg3 380–394, Dsg3 763–777, Dsg3 810–824 and Dsg3 963–977 (numbered in accordance with Swiss-Prot [26] accession number P32926), capable of stimulating patient derived T cells, were selected. The binding of these peptides to the DR and DQ structural models were studied by our efficient computational docking protocol [27]. In the light shed by these atomic models, the binding specificities of each allele to the various Dsg3 peptides are discussed. The results obtained in the study are able to discriminate between PV associated and non-associated alleles, consistent with the experimental results obtained by Veldman et al. [28] and Sinha et al. [unpublished results for Dsg3 342–356, 810–824 and 963–977]. Insights into structural features behind the immune response provided by protective alleles for PV have also been obtained by our structural immunoinformatics approach.
Results and discussion
Allele comparisons – HLA DR4 PV
Sequence and structural similarity between the eight (DRB1*0402, *0404, *0406, *1401, *1404, *1405, DQB1*0202, and *0503) MHC structural models and their corresponding template structures (1D5Z: DRB1*0401, 1S9V: DQB1*0201, 1UVQ: DQB1*0602). Positives represent a measure of sequence similarity, accounting for identical and conservatively substituted residues. Root mean square deviations (RMSD) values in Å are shown for the Cα atoms of both MHC chains and for the residues comprising the different peptide-binding pockets.
|
Allele |
Template |
Sequence Identity |
Positives |
Cα RMSD (Å) |
|||||
|---|---|---|---|---|---|---|---|---|---|
|
α & β chains |
Pockets |
||||||||
|
P1 |
P4 |
P6 |
P7 |
P9 |
|||||
|
DRB1*0402 |
1D5Z |
97.9% |
99.0% |
0.35 |
0.12 |
0.06 |
0.07 |
0.10 |
0.09 |
|
DRB1*0404 |
1D5Z |
99.0% |
99.5% |
0.31 |
0.15 |
0.10 |
0.06 |
0.07 |
0.18 |
|
DRB1*0406 |
1D5Z |
97.9% |
98.4% |
0.32 |
0.11 |
0.15 |
0.07 |
0.11 |
0.22 |
|
DRB1*1401 |
1D5Z |
94.1% |
97.3% |
0.25 |
0.11 |
0.09 |
0.02 |
0.09 |
0.18 |
|
DRB1*1404 |
1D5Z |
85.8% |
89.5% |
0.29 |
0.12 |
0.10 |
0.02 |
0.06 |
0.22 |
|
DRB1*1405 |
1D5Z |
81.0% |
83.2% |
0.24 |
0.11 |
0.07 |
0.02 |
0.08 |
0.07 |
|
DQB1*0202 |
1S9V |
98.0% |
99.0% |
0.57 |
0.16 |
0.09 |
0.04 |
0.15 |
0.05 |
|
DQB1*0503 |
1UVQ |
93.0% |
96.0% |
0.39 |
0.03 |
0.07 |
0.01 |
0.10 |
0.06 |

Multiple sequence alignment of the MHC DR and DQ alleles β chain. Pocket residues are shaded in black.
Allele comparisons – HLA DR6 PV
Study of individual allele frequencies in DR6 PV patients revealed that the relevant disease susceptibility allele is DQB1*0503 instead of DR6 alleles [Sinha et al., manuscript in preparation]. DQB1*0503 and the DR6 PV non-associated alleles investigated in this study show a significant degree of overlap in alignment, with 14 amino acid differences in areas of the binding cleft that could affect peptide binding. Clear differences in the amino acid sequences are observed at residue β86 of pocket 1, residues β13, β70, β71, β74, β78 of pocket 4, residue β11 of pocket 6, residues β28, β30, β67, β71 of pocket 7 and residues β9, β37, β57, β60 of pocket 9. Similar to the DR4 alleles, all five important peptide-binding pockets 1, 4, 6, 7 and 9 in DBQ1*0503 and DR6 alleles demonstrate exceptionally high structural conservation at the Cα positions. A significant difference is that DQB1*0503 contains a negatively charged Asp β57 that differs from the uncharged Ala β57 found in non-PV associated DRB1*1401 and *1404. Also, at positions β70 and β71, DQB1*0503 does not contain negatively charged residues identified in DRB1*0402 that are critical for binding of self-antigens in DR4 PV patients. Instead, these positions were replaced by two small neutral hydrophobic residues (Gly β70 and Ala β71), suggesting that DRB1*0402 and DQB1*0503 may recognize different sets of PV epitopes under the influence of a different balance of intermolecular forces. Positions β70 and β74 show charge reversal, in the non-PV associated DRB1*1401, *1404 and *1405 alleles while the negative charge at β71 alone is conserved, compared to DRB1*0402, making pocket 4 the single dominant factor discriminating between PV non-association and susceptibility.
Allele comparisons – PV protective and susceptible alleles
Differences in the amino acid sequences are observed at residue β86 of pocket 1, residue β70, β71 of pocket 4, residues β28, β30, β47, β71 of pocket 7 and residues β37, β57 of pocket 9. Both protective alleles (DQB1*0201 and DQB1*0202) do not contain negatively charged residues at position β70 (pocket 4) and β71 (pocket 7). Instead, these positions were replaced by two large and positively charged amino acids (Arg β70 and Lys β71). The functional specificities of PV protective and susceptible alleles are also affected by clear structural differences in the Cα positions of both α and β chains (Cα RMSD > 0.57Å) indicating that any differences in peptide discrimination between the alleles is due to a combination of both the backbone conformation as well as the size and nature of the side chains of the pocket residues.
Epitope comparisons – HLA DR4 PV
Eight previously identified stimulatory Dsg3 epitopes (Dsg3 191–205, Dsg3 206–220, Dsg3 252–266, Dsg3 342–356, Dsg3 380–394, Dsg3 763–777, Dsg3 810–824 and Dsg3 963–977) for DRB1*0402 were docked into the binding groove of all DR4 (DRB1*0401, *0402, *0404, *0406) alleles investigated in this study. Analysis of these Dsg3 peptide-bound alleles revealed that only one peptide conformation can fit perfectly into the binding cleft of DRB1*0402, and atomic clashes of these Dsg3 peptides are obtained for all other DR4 subtypes investigated in this study. Notably, two epitopes (Dsg3 342–356 and Dsg3 810–824) have small residues (Ser/Cys) in pocket 1, suggesting that large anchor residues may play a critical role for high affinity binding in DR4 PV molecules, an observation previously documented for influenza-associated I-Ad allele of mice [31]. This finding provides support to the evidence that DRB1*0402 is associated with PV whereas other DR4 subtypes are non-associated, with the exception of DRB1*0406 that is reported to be associated in the Japanese population [32]. As such, there is a possibility of the existence of other peptides relevant in the Japanese populations that bind to *0406 but are yet to be determined.
Epitope comparisons – HLA DR6 PV
Dsg3 96–112, a recently identified epitope in DR6 PV patients [28], fits perfectly into the binding groove of DQB1*0503 with two identified core sequences at residues 101–109 and residues 102–110. The identified 101–109 core has four intermolecular hydrogen bonds compared to seven intermolecular hydrogen bonds in the core of 102–110. Perfect fitting of Dsg3 206–220, Dsg3 252–266, Dsg3 342–356, Dsg3 810–824 and Dsg3 963–977 into the binding groove of DQB1*0503 is also obtained. Atomic clashes are obtained for Dsg3 191–205, Dsg3 380–394 and Dsg3 763–777 as well as all DR6 alleles investigated in this study. The proportion of DRB1*1401, *1404 and *1405 has been reported to be increased in PV probably due to linkage disequilibrium. The lack of binding of all stimulatory peptides investigated in this study to these alleles indicates that the HLA association in DR6 PV patients is more likely at the DQB1 locus (DQB1*0503 allele) and not the linked DRB1 loci (DRB1*1401, *1404 and *1405). Our data supports the notion that the reported associations of this disease with DRB1*1401, *1404, *1405 are due to linkage disequilibrium with the true disease associated allele (DQB1*0503).
Epitope comparisons – PV susceptibility Alleles
Preferred core residues for PV associated alleles. Best fitting nonameric core residues in the binding groove are underlined.
|
No. |
Residues |
Allele |
Core peptide sequences |
No. |
Residues |
Allele |
Core peptide sequences |
|---|---|---|---|---|---|---|---|
|
I |
96–112 |
DRB1*0402 |
- |
V |
342–356 |
DRB1*0402 |
SVKLSIAVKNKAEFH |
|
DQB1*0503 |
PFGIFVVDKNTGDINIT |
DQB1*0503 |
SVKLSIAVKNKAEFH |
||||
|
PFGIFVVDKNTGDINIT |
VI |
380–394 |
DRB1*0402 |
GIAFRPASKTFTVQK |
|||
|
II |
191–205 |
DRB1*0402 |
NSKIAFKIVSQEPAG |
DQB1*0503 |
--- |
||
|
DQB1*0503 |
--- |
VII |
763–777 |
DRB1*0402 |
SGTMRTRHSTGGTNK |
||
|
III |
206–220 |
DRB1*0402 |
TPMFLLSRNTGEVRT |
DQB1*0503 |
--- |
||
|
DQB1*0503 |
TPMFLLSRNTGEVRT |
VIII |
810–824 |
DRB1*0402 |
NDCLLIYDNEGADAT |
||
|
IV |
252–266 |
DRB1*0402 |
ECNIKVKDVNDNFPM |
DQB1*0503 |
NDCLLIYDNEGADAT |
||
|
DQB1*0503 |
ECNIKVKDVNDNFPM |
IX |
963–977 |
DRB1*0402 |
ERVICPISSVPGNLA |
||
|
ECNIKVKDVNDNFPM |
DQB1*0503 |
ERVICPISSVPGNLA |
Epitope comparisons – PV protective alleles
Our simulation results indicate that DQB1*0201 and DQB1*0202 can bind to multiple core sequences for the majority of PV epitopes investigated in this study. DQB1*0201 can bind one epitope (Dsg3 963–977) at two core regions, one epitope (Dsg3 206–220) at three core regions, three epitopes (Dsg3 191–205, 252–266 and 342–356) at four core regions, and two epitopes (Dsg3 96–112 and 810–824) at five core regions. DQB1*0202 can bind two epitopes (Dsg3 96–112 and 963–977) at three core regions, two epitopes (Dsg3 342–356 and 810–824) at four core regions and one epitope (Dsg3 252–266) at five core regions. In contrast, the majority of PV epitopes (with the exception of Dsg3 96–112 and 252–266) can bind to PV susceptible alleles DRB1*0402 and DQB1*0503 at a single core. This finding lends support to the hypothesis that the protective alleles DQB1*0201, *0202 may be capable of binding to most peptides with greater affinity than PV susceptible alleles, allowing for efficient deletion of autoreactive T cells [33].
Role of flanking residues in peptide selection
Our data demonstrates that the conformations of flanking peptide residues that extend beyond the binding groove are critical to peptide selection in MHC class II alleles. The core sequences of Dsg3 963–977 fit perfectly within the binding grooves of non-associated alleles DRB1*0401, *0404, and *1404 but poor contacts to the respective alleles at Phe α50 are obtained when the conformation of the N-terminal flanking residue Ile4 is taken into account. These results suggest that binding is determined by both the core and flanking segments while considering the overall interactions between each peptide and the respective alleles.
Sequence motifs
Comparison of core peptides (numbering according to Table 2) from structural docking in the different binding pockets with the sequence-based binding motifs. '+' indicates compliance of amino acid residues within the core (bold underlined) with the respective binding motifs defined by the groups of a Veldman [28] and bSinha [2, unpublished results].
|
No. |
Residues |
Peptide Sequence and positions in the bound conformation for DRB1*0402 |
Core peptide residue positions as defined by binding motifs |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
p1 |
p4 |
p6 |
p9 |
|||||||||
|
1 2 3 4 5 6 7 8 9 |
V a |
S b |
V |
S |
V |
S |
V |
S |
||||
|
II |
191–205 |
NSKIA |
F K I V S Q E P A |
G |
+ |
+ |
+ |
+ |
||||
|
III |
206–220 |
TPM |
F L L S R N T G E |
VRT |
+ |
+ |
+ |
+ |
||||
|
IV |
252–266 |
ECNI |
K V K D V N D N F |
PM |
+ |
+ |
||||||
|
V |
342–356 |
SVKL |
S I A V K N K A E |
FH |
+ |
+ |
+ |
+ |
||||
|
VI |
380–394 |
GIA |
F R P A S K T F T |
VQK |
+ |
+ |
+ |
+ |
||||
|
VII |
763–777 |
SGT |
M R T R H S T G G |
TNK |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
|
VIII |
810–824 |
ND |
C L L I Y D N E G |
ADAT |
+ |
+ |
+ |
|||||
|
IX |
963–977 |
ERVICP |
I S S V P G N L A |
+ |
+ |
+ |
+ |
|||||
Peptide VII (Dsg3 763–777) agrees well with the motifs from Veldman et al. [28] and Sinha et al. (unpublished results), while all other peptides show low to moderate compliance. Of the four positions compared, peptide IV (Dsg3 252–266) shows agreement only at position p6. For Dsg3 342–356 peptide, the core nonamer identified by our models is 346–354, which is register-shifted by one residue from the core of 347–355 reported by Veldman et al. [28], and 345–353 identified by Sinha et al. (unpublished results), for the binding groove of *0402. This shift is critical as residues p1 and p4 identified by us do not fit well into both binding motifs. Our modeling studies suggest that peptide position p4 need not be positively charged as indicated by Veldman et al. [28], supporting the existence of a more degenerate motif by Sinha et al. (unpublished results) at this position. In addition, p1 also appears to be more degenerate than previously suggested [28], showing a preference for hydrophobic and large residues but can accommodate residues of other sizes as well. Hence for generating sequence patterns to design peptides for vaccine design, structural information is important [34] and the exact peptide in the binding groove identified by our docking protocol will be most useful here.
Disease progression in PV
T cell response to a number of epitopes among PV patients has been reported in several studies [2, 8, 26, 29, 30]. There may be disease heterogeneity, meaning that clinically similar but distinct phenotypes could operate by alternate pathways, each with a different initial immunodominant epitope(s). The differential T cell reactivities among individual patients to individual peptides may also be a function of the disease stage or severity and correlate with mechanisms of disease progression. While there may be a limited set of epitopes present in patients in the early stages of the disease, epitope spreading can occur during disease progression, resulting in reactivity to previously innocuous epitopes. In addition, reactivities to multiple epitopes within individual patients were detected in two cases (Dsg3 191–205 and 342–356 for PV107; Dsg3 191–205, 810–824 and 963–977 for PV117). Autoantibodies against desmoglein 1 have also been reported in severe disease [35]. One other incidence of multiple T cell reactivities within a PV patient has been previously reported [29]. These findings, together with our simulation results, lend further credence to the hypothesis that no single epitope is responsible for both disease initiation and propagation and are consistent with the expected and observed ability to generate multiple pMHC complexes from a single target autoantigen.
Conclusion
Docking simulations at the binding site of PV associated and non-associated DR and DQ alleles have been performed to analyze the structural aspects of binding and allele-specificity for nine previously identified Dsg3 epitopes. To represent the possibility that any core peptide sequences can be recognized by the binding groove of MHC class II alleles, a sliding window was applied to generate all possible combinations of core nonamer peptides from each Dsg3 peptide. This method can eliminate any bias in selecting core peptides based on sequence patterns alone.
We have found the existence of best-fit core residues at different positions of each peptide (excepting Dsg3 96–112) into the binding groove of DRB1*0402 with no observed atomic clash penalties or bad contacts. In contrast, atomic clashes are experienced in all other PV non-associated DR4 alleles. This discrimination further establishes the crucial role that DRB1*0402 plays in selecting specific self-peptides in DR4 PV. In addition, we found that DRB1*0402 and DQB1*0503 do not necessarily share the same core residues. It is possible that DRB1*0402, DQB1*0503 and all other PV non-associated alleles may have different sets of binding specificities. Our studies also indicate that perfect fitting of the core nonameric peptide residues within the binding groove of MHC class II alleles may not guarantee perfect fitting of the entire peptide, and flanking residues outside the binding groove may play a critical part in peptide selection. Such binding interactions suggest that longer peptides extending out of the binding groove of MHC class II alleles must be taken into account in the generation of HLA class II binding motifs and for vaccine design.
The comparison of core peptide residues with binding motifs previously defined by Veldman et al. [28] and Sinha et al. (unpublished results) indicates that sequence-based methods are currently insufficient for the design of PV epitopes as there are both register shifts in the suggested motifs as well as polymorphism observed in the core residues in the binding groove. More experimental data are necessary for the definition of DR4 and DR6 PV specific binding motifs.
The methodology presented here may serve as a general method suitable for finding allelic specific peptides, applicable to the design of both sub-type specific vaccines as well as promiscuous peptide epitopes. In particular, this approach is useful in situations where there is insufficient data for training sequence-based predictive models. In the context of PV, this approach provides a means for discriminating between peptide binders and non-binders for a number of PV implicated alleles where training data is deficient. Our results support the hypothesis [33] that the alleles DQB1*0201 and *0202 play a protective role by binding Dsg3 peptides with greater affinity than the susceptible alleles, facilitating efficient deletion of autoreactive T cells. With increasing evidence indicating that no single epitope may be responsible for both disease initiation and propagation in PV, it is valuable to identify all Dsg3 peptides that bind to the PV susceptible alleles. Specifically, the identification of peptides that bind to both DRB1*0402 and DQB1*0503 are of great importance as these peptides may serve as targets for epitope-based therapeutic vaccination of both DR4 and DR6 PV patients. Future work will include autoantigens from Dsg1, the main causative agent for pemphigus foliaceus and reported in severe cases of PV.
Materials and methods
Template search
In this study, ten PV associated, closely related non-associated and protective MHC class II alleles DRB1*0401, *0402, *0404, *0406, *1401, *1404, *1405, DQB1*0201, *0202, and *0503 were selected for analysis. MHC sequence data were obtained from IMGT-HLA http://www.ebi.ac.uk/imgt/hla/[36] database. The α chain of all DR alleles investigated in this study is the DRA1*0101 sequence, with the β chain from the allele sub-type. To identify potential structural templates available in the Protein Data Bank (PDB) [37] for model building, a sequence similarity search was performed using PSI-BLAST [38] running on the servers at NCBI http://www.ncbi.nlm.nih.gov/blast/ and the highest quality templates were selected among the returned results. Among these, the crystal structures of HLA-DR4 (PDB code 1D5Z) and HLA-DQ2 (PDB code 1S9V) were adopted as the structures of DRB1*0401 and DQB1*0201 respectively (100% sequence identity). The crystal structures of DRB1*0401 (PDB code 1D5Z), DQB1*0602 (PDB code 1UVQ) and DQB1*0201 (PDB code 1S9V) were selected as templates for all other DR subtypes, DQB1*0503 and DQB1*0202 respectively (Table 1).
Model building
The program MODELLER [39] was employed for comparative modeling of both DRB1 (*0402, *0404, *0406, *1401, *1404, *1405) and DQB1 (*0202, *0503) subtypes. The models are constructed by optimally satisfying spatial constraints obtained from the alignment of the template structure with the target sequence and from the CHARMM-22 force field [40]. The initial model was refined by assigning the rotameric states of essential side chains according to the corresponding crystal structure, followed by a short energy minimization [41] from the program Internal Coordinates Mechanics (ICM; Molsoft LLC, San Diego, CA) [42].
Patient recruitment and groupings
HLA class II haplotypes of PV patients (PV) and controls (CR). Typing was performed at the Rogosin Institute, New York Presbyterian Hospital, NY.
|
Subject |
Sex |
Age |
DRB1 |
DQB1 |
DQA |
|---|---|---|---|---|---|
|
PV104 |
F |
64 |
*0402, *0403 |
*0302, *0304 |
*03011 |
|
PV105 |
F |
73 |
*1404, *0102 |
*0503, *0501 |
*01041, *0101 |
|
PV107 |
F |
70 |
*0402, *0102 |
*0302, *0501 |
*03011, *0101 |
|
PV108 |
F |
52 |
*0402, *0404 |
*0302, *0304 |
*03011, *0303 |
|
PV112 |
F |
57 |
*0402, *1101 |
*0302, *0301 |
*03011, *0505 |
|
PV114 |
M |
50 |
*0402, *1401 |
*0503, *0302 |
*03011, *01041 |
|
PV115 |
F |
34 |
*0402, *0701 |
*0302, *0202 |
*03011, *0201 |
|
PV117 |
M |
44 |
*0402, *0403 |
*0302, *0305 |
*03011 |
|
PV118 |
M |
62 |
*0402, *1302 |
*0601, *0402 |
*0102, *0303 |
|
CR101 |
F |
58 |
*0402, *0701 |
*0202, *0302 |
*0201, *0301 |
|
CR102 |
M |
81 |
*0402, *1101 |
*0301, *0302 |
*0501-05, *0301 |
Peptide set
Nine previously identified epitopes Dsg3 96–112, 191–205, Dsg3 206–220, Dsg3 252–266, Dsg3 342–356, Dsg3 380–394, Dsg3 763–777, Dsg3 810–824 and Dsg3 963–977 that elicited primary proliferative T cell response in PV patients [2, 8, 26, 29, 30] were selected for modeling studies. T cell response to eight of these peptides (Dsg3 191–205, Dsg3 206–220, Dsg3 252–266, Dsg3 342–356, Dsg3 380–394, Dsg3 763–777, Dsg3 810–824 and Dsg3 963–977) has been reported in patients carrying DRB1*0402. Dsg3 96–112 has been reported to elicit T cell response in patients with DQB1*0503 but lacking DRB1*0402 [28]. Of these Dsg3 191–205, Dsg3 342–356, Dsg3 810–824 and Dsg3 963–977 were shown to directly bind to the DRB1*0402 receptor by competitive binding assays (Sinha et al., unpublished results). Briefly, soluble HLA DRA1*0101/DRB1*0402 were purified by DR-specific affinity chromatography and incubated with different concentrations of experimental peptides (0–40 μM) in the presence of biotinylated class II-associated invariant-chain peptide (CLIP) (1 μM) for 2 hours. The MHC-peptide complexes were then captured on a 96-well plate coated with anti-HLA-DR (L243) (BD Pharmingen, San Diego, CA). The CLIP bound to the MHC molecules was directly assayed using Europium (Eu)-labeled streptavidin (Perkin Elmer, Boston, MA). The relative binding of peptides was subsequently determined by measuring the displacement of the CLIP at different peptide concentrations.
Peptide docking
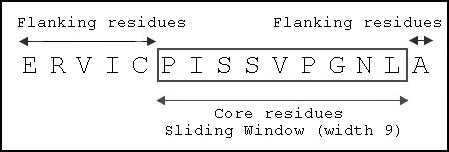
Sliding window of width 9 applied to identify core residues of Dsg3 963–977 to be modeled into binding groove.
Definition of contact residues
In this study, MHC-peptide residues were considered to be in contact if at least one pair of their non-hydrogen ("heavy") atoms was found to be within 4.00Å radius [47]. Intra-peptide interactions and intra-MHC interactions were not considered as they have minor influence on peptide/protein backbone structure. Any atom in the peptide and any atom in the MHC were considered to be experiencing atomic clash if their separation is below 2.00Å [48] for non-hydrogen atoms and below 1.60Å for atoms participating in hydrogen bonds [49, 50].
Definition of binding pocket for MHC Class II alleles
Interactions between side-chains of bound peptide ligands and polymorphic cavities (or anchor "pockets") in the binding site of MHC class II alleles are important in determining the peptide binding affinity and sequence specificity of MHC molecules and are defined according to the work of Stern et al. [51, 52].
List of abbreviations
- PV:
-
Pemphigus vulgaris
- Dsg3:
-
Desmoglein-3
- MHC:
-
Major histocompatibility complex
- HLA:
-
Human leukocyte antigen
Declarations
Authors’ Affiliations
References
- Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL: Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med 1993, 178:27–47.View ArticlePubMedGoogle Scholar
- Sinha AA, Lopez MT, McDevitt HO: Autoimmune diseases: the failure of self-tolerance. Science 1990, 248:1380–1388.View ArticlePubMedGoogle Scholar
- Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, McDermott M, Sinha AA, Timmerman L, Steinman L, et al.: A molecular basis for MHC class II-associated autoimmunity. Science 1988, 240:1003–1009.View ArticlePubMedGoogle Scholar
- Amagai M: Autoantibodies against cell adhesion molecules in pemphigus. J Dermatol 1994, 21:833–837.PubMedGoogle Scholar
- Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, Alper CA, Yunis EJ: Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci USA 1991, 88:5056–5060.View ArticlePubMedGoogle Scholar
- Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, Awdeh Z, Alper CA: Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci USA 1990, 87:7658–7662.View ArticlePubMedGoogle Scholar
- Scharf SJ, Freidmann A, Steinman L, Brautbar C, Erlich HA: Specific HLA-DQB and HLA-DRB1 alleles confer susceptibility to pemphigus vulgaris. Proc Natl Acad Sci USA 1989, 86:6215–6219.View ArticlePubMedGoogle Scholar
- Sinha AA, Brautbar C, Szafer F, Friedmann A, Tzfoni E, Todd JA, Steinman L, McDevitt HO: A newly characterized HLA DQ beta allele associated with pemphigus vulgaris. Science 1988, 239:1026–1029.View ArticlePubMedGoogle Scholar
- Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A: Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol 1998, 160:3363–3373.PubMedGoogle Scholar
- Mallios RR: Class II MHC quantitative binding motifs derived from a large molecular database with a versatile iterative stepwise discriminant analysis meta-algorithm. Bioinformatics 1999, 15:432–439.View ArticlePubMedGoogle Scholar
- Mallios RR: Predicting class II MHC/peptide multi-level binding with an iterative stepwise discriminant analysis meta-algorithm. Bioinformatics 2001, 17:942–948.View ArticlePubMedGoogle Scholar
- Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L: Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. Bioinformatics 1998, 14:121–130.View ArticlePubMedGoogle Scholar
- Noguchi H, Hanai T, Honda H, Harrison LC, Kobayashi T: Fuzzy neural network-based prediction of the motif for MHC class II binding peptides. J Biosci Bioeng 2001, 92:227–231.View ArticlePubMedGoogle Scholar
- Noguchi H, Kato R, Hanai T, Matsubara Y, Honda H, Brusic V, Kobayashi T: Hidden Markov model-based prediction of antigenic peptides that interact with MHC class II molecules. J Biosci Bioeng 2002, 94:264–270.PubMedGoogle Scholar
- Hammer J, Bono E, Gallazzi F, Belunis C, Nagy Z, Sinigaglia F: Precise prediction of major histocompatibility complex class II-peptide interaction based on peptide side chain scanning. J Exp Med 1994, 180:2353–2358.View ArticlePubMedGoogle Scholar
- Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O: Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics 2004, 20:1388–1397.View ArticlePubMedGoogle Scholar
- Karpenko O, Shi J, Dai Y: Prediction of MHC class II binders using the ant colony search strategy. Artif Intell Med 2005, 35:147–156.View ArticlePubMedGoogle Scholar
- Doytchinova IA, Flower DR: Towards the in silico identification of class II restricted T-cell epitopes: a partial least squares iterative self-consistent algorithm for affinity prediction. Bioinformatics 2003, 19:2263–2270.View ArticlePubMedGoogle Scholar
- Logean A, Rognan D: Recovery of known T-cell epitopes by computational scanning of a viral genome. J Comput Aided Mol Des 2002, 16:229–243.View ArticlePubMedGoogle Scholar
- Altuvia Y, Schueler O, Margalit H: Ranking potential binding peptides to MHC molecules by a computational threading approach. J Mol Biol 1995, 249:244–250.View ArticlePubMedGoogle Scholar
- Lim SK, Kim S, Lee HG, Lee KY, Kwon TJ, Kim K: Selection of peptides that bind to the HLA-A2.1 molecule by molecular modeling. Mol Immunol 1996, 33:221–230.View ArticlePubMedGoogle Scholar
- Michielin O, Luescher I, Karplus M: Modeling the TCR-MHC-peptide complex. J Mol Biol 2000, 300:1205–1235.View ArticlePubMedGoogle Scholar
- Michielin O, Karplus M: Binding free energy differences in a TCR-peptide-MHC complex induced by a peptide mutation: a stimulation analysis. J Mol Biol 2002, 324:547–569.View ArticlePubMedGoogle Scholar
- Doytchinova IA, Guan P, Flower DR: Identifying human MHC supertypes using bioinformatic methods. J Immunol 2004, 172:4314–4323.PubMedGoogle Scholar
- Doytchinova IA, Flower DR:In silico identification of supertypes for class II MHCs. J Immunol 2005, 174:7085–7095.PubMedGoogle Scholar
- Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M: The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 2003, 31:365–370.View ArticlePubMedGoogle Scholar
- Tong JC, Tan TW, Ranganathan S: Modeling the structure of bound peptide ligands to major histocompatibility complex. Protein Sci 2004, 13:2523–2532.View ArticlePubMedGoogle Scholar
- Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grötzinger J, Schultz E, Hertl M: T cell recognition of Desmoglein 3 peptides in patients with Pemphigus Vulgaris and healthy individuals. J Immunol 2003, 172:3883–3892.Google Scholar
- Wucherpfennig KW, Yu B, Bhol K, Monos DS, Argyris E, Karr RW, Ahmed AR, Strominger JL: Structural basis for major histocompatibility complex (MHC)-linked susceptibility to autoimmunity: charged residues of a single MHC binding pocket confer selective presentation of self-peptides in pemphigus vulgaris. Proc Natl Acad Sci USA 1995, 92:11935–11939.View ArticlePubMedGoogle Scholar
- Hertl M, Karr RW, Amagai M, Katz SI: Heterogeneous MHC II restriction pattern of autoreactive desmoglein 3 specific T cell responses in pemphigus vulgaris patients and normals. J Invest Dermatol 1998, 110:388–392.View ArticlePubMedGoogle Scholar
- Scott CA, Peterson PA, Teyton L, Wilson IA: Crystal structures of two I-A d -peptide complexes reveal that high affinity can be achieved without large anchor residues. Immunity 1998, 8:319–329.View ArticlePubMedGoogle Scholar
- Yamashina Y, Miyagawa S, Kawatsu T, Iida T, Higashimine I, Shirai T, Kaneshige T: Polymorphisms of HLA class II genes in Japanese patients with pemphigus vulgaris. Tissue Antigens 1998, 52:74–77.View ArticlePubMedGoogle Scholar
- Gebe JA, Swanson E, Kwok WW: HLA class II peptide-binding and autoimmunity. Tissue Antigens 2002, 59:78–87.View ArticlePubMedGoogle Scholar
- Schirle M, Weinschenk T, Stevanovic S: Combining computer algorithms with experimental approaches permits the rapid and accurate identification of T cell epitopes from derived antigens. J Immunol Methods 2001, 257:1–16.View ArticlePubMedGoogle Scholar
- Harman KE, Gratian MJ, Bhogal BS, Challacombe SJ, Black MM: A study of desmoglein 1 autoantibodies in pemphigus vulgaris: Racial differences in frequency and the association with a more severe phenotype. Br J Dermatol 2000, 143:343–348.View ArticlePubMedGoogle Scholar
- Robinson J, Waller MJ, Parham P, de Groot N, Bontrop R, Kennedy LJ, Stoehr P, Marsh SGE: IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocompatibility complex. Nucleic Acids Res 2003, 31:311–314.View ArticlePubMedGoogle Scholar
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Res 2000, 28:235–242.View ArticlePubMedGoogle Scholar
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215:403–410.PubMedGoogle Scholar
- Sali A, Blundell TL: Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol 1993, 234:779–815.View ArticlePubMedGoogle Scholar
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al.: All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem 1998, B102:3586–3617.Google Scholar
- Abagyan R, Totrov M, Kuznetsov D: ICM – a new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comp Chem 1994, 15:488–506.View ArticleGoogle Scholar
- Abagyan R, Totrov M: Ab Initio Folding of Peptides by the Optimal-Bias Monte Carlo Minimization Procedure. J Comput Phys 1999, 151:402–421.View ArticleGoogle Scholar
- Rammennsee HG, Friede T, Stevanovic S: MHC ligands and peptide motifs: first listing. Immunogenetics 1995, 41:178–228.View ArticleGoogle Scholar
- Totrov M, Abagyan R: Protein-ligand docking as an energy optimization problem. Drug-receptor thermodynamics: Introduction and experimental applications (Edited by: Raffa RB). New York: John Wiley & Sons 2001, 603–624.Google Scholar
- Totrov M, Abagyan R: Derivation of sensitive discrimination potential for virtual ligand screening. Proceedings of the Third Annual International Conference on Computational Molecular Biology Lyon, France: ACM Press 1999, 37–38.Google Scholar
- Cavasotto CN, Orry AJW, Abagyan R: Structure-based identification of binding sites, native ligands and potential inhibitors for G-protein coupled receptors. Proteins 2003, 51:423–433.View ArticlePubMedGoogle Scholar
- Fischer KF, Marquesee S: A rapid test for identification of autonomous folding units in proteins. J Mol Biol 2000, 302:701–712.View ArticlePubMedGoogle Scholar
- Samudrala R, Moult J: Handling Context-Sensitivity in Protein Structures usinf graph theory: Bona Fide Prediction. Proteins 1997, (Suppl 1):43–49.Google Scholar
- Wallace AC, Laskowski RA, Thornton JM: LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Prot Eng 1995, 8:127–134.View ArticleGoogle Scholar
- Samanta U, Bahadur RP, Chakrabarti P: Quantifying the accessible surface area of protein residues in their local environment. Prot Eng 2002, 15:659–667.View ArticleGoogle Scholar
- Stern LJ, Wiley DC: Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 1994, 2:245–251.View ArticlePubMedGoogle Scholar
- Murthy VL, Stern LJ: The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure 1997, 5:1385–1396.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.