

Ribosomal protein mRNAs are translationally-regulated during human dendritic cells activation by LPS
- Research
- Open Access
Ribosomal protein mRNAs are translationally-regulated during human dendritic cells activation by LPS
- Maurizio Ceppi1_32, 2_32, 3_32, 6_32,
- Giovanna Clavarino1_32, 2_32, 3_32,
- Evelina Gatti1_32, 2_32, 3_32,
- Enrico K Schmidt1_32, 2_32, 3_32,
- Aude de Gassart1_32, 2_32, 3_32,
- Derek Blankenship5_32,
- Gerald Ogola5_32,
- Jacques Banchereau4_32,
- Damien Chaussabel4_32 and
- Philippe Pierre1_32, 2_32, 3_32Email author
- Received: 30 September 2009
- Accepted: 27 November 2009
- Published: 27 November 2009
Abstract
Background
Dendritic cells (DCs) are the sentinels of the mammalian immune system, characterized by a complex maturation process driven by pathogen detection. Although multiple studies have described the analysis of activated DCs by transcriptional profiling, recent findings indicate that mRNAs are also regulated at the translational level. A systematic analysis of the mRNAs being translationally regulated at various stages of DC activation was performed using translational profiling, which combines sucrose gradient fractionation of polysomal-bound mRNAs with DNA microarray analysis.
Results
Total and polysomal-bound mRNA populations purified from immature, 4 h and 16 h LPS-stimulated human monocyte-derived DCs were analyzed on Affymetrix microarrays U133 2.0. A group of 375 transcripts was identified as translationally regulated during DC-activation. In addition to several biochemical pathways related to immunity, the most statistically relevant biological function identified among the translationally regulated mRNAs was protein biosynthesis itself. We singled-out a cluster of 11 large ribosome proteins mRNAs, which are disengaged from polysomes at late time of maturation, suggesting the existence of a negative feedback loop regulating translation in DCs and linking ribosomal proteins to immuno-modulatory function.
Conclusion
Our observations highlight the importance of translation regulation during the immune response, and may favor the identification of novel protein networks relevant for immunity. Our study also provides information on the potential absence of correlation between gene expression and protein production for specific mRNA molecules present in DCs.
Keywords
- Ingenuity Pathway Analysis
- Translation Regulation
- Murine Double Minute
- Systemic Onset Juvenile Idiopathic Arthritis
- Sucrose Gradient Fractionation
Background
Dendritic cells (DCs) are haematopoietic cells specialized in antigen capture and presentation for initiation of primary and secondary immune responses. Due to this central role in induction and regulation of immunity, they represent an attractive target for immunotherapy against various diseases, including cancer and microbial infections [1]. We recently demonstrated that translation regulation is required for function and survival of mouse activated DCs [2]. Moreover, emerging evidence indicate that translation plays a major role in immune regulation and its dysfunction can lead to pathology [3, 4, 5]. Although several seminal studies have described the use of microarrays to define the gene expression and functional signature of DCs upon pathogen detection [6, 7], there were no attempts to include the additional layer of complexity brought by translational regulation. As the relationship between inflammation, innate immunity, and post-transcriptional regulation is becoming clearer [8], we have in a recent study used a microarray-based screen to identify the immunologically relevant pathways regulated by miR-155 in lipopolysaccharide (LPS)-activated human monocyte-derived DC (moDC) [9]. To increase further our understanding of post-transcriptional regulation and establish the contribution of translation in the control of immune response, we carried-out, using Affymetrix microarrays, a systematic and comparative analysis of polysome-bound mRNA [10, 11, 12] purified from differently LPS-activated moDCs.
Using this approach, and in addition to several immunologically relevant mRNAs, we identified a network of ribosomal protein mRNAs being strongly down-modulated at the translational level at late time of DC maturation. Ribosomal proteins are integral components of the basal cellular machinery involved in protein synthesis, whose roles have been regarded collectively as important, but individually disregarded. Recent findings, however, have demonstrated that components of the translational apparatus are multifunctional and that several individual ribosomal proteins play a role in regulating cell growth, transformation and death [13]. Our results clearly support these views and underline the importance of these proteins for DC function.
Results and discussion
Translation is regulated in LPS-activated human moDCs
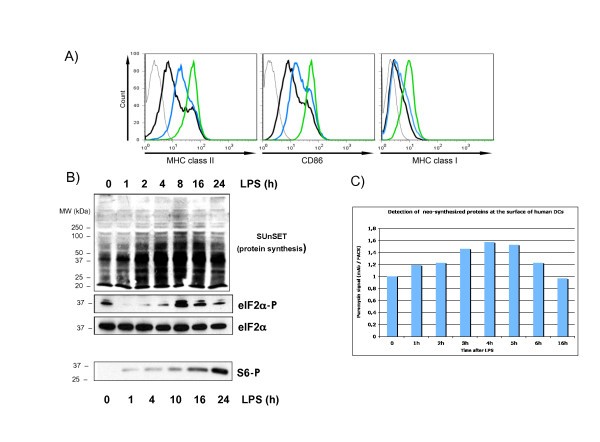
Translation is regulated in LPS-activated moDCs. moDCs were stimulated with LPS for the indicated timepoints and after harvesting both the maturation status and protein synthesis were monitored in parallel. (A) The surface accumulation of MHC II, CD86 and MHC I, were measured by flow cytometry in immature (black line), 4 h (blue) or 16 h (green) LPS-stimulated moDCs. (B) The rate of protein synthesis was monitored with puromycin incorporation using immunoblots (top). Cells extracts were separated by denaturing electrophoresis and analyzed by western blot with antibody to the phosphorylated form of eIF2α and the S6 ribosomal protein (bottom). Total eIF2α detection is shown for equal loading control. (C) FACS analysis with antibody to puromycin (SUnSET method) to quantify protein synthesis. Data are representative of at least three independent experiments, each derived from a different DC preparation.
Translation initiation is also controlled through the stimulation of the PI3 kinase/AKT/mTOR signal transduction cascade, which leads to the phosphorylation of eIF4E-binding proteins (4E-BPs) and of the S6 ribosomal protein (S6) by the ribosomal protein S6 kinase (S6K1) [16]. Both molecules have an important role in regulating cap-mediated translation and S6 phosphorylation represents a hallmark of protein synthesis activation. S6 phosphorylation was monitored in LPS-activated moDCs by immunoblot (Figure 1B). We confirmed that S6 phosphorylation is steadily and strongly increased over time of LPS-exposure and this in a correlated manner to the moDCs maturation phenotype.
Thus, protein synthesis is tightly controlled upon LPS-sensing by MoDCs and is likely to represent a functionally important aspect of human DC maturation. Furthermore, this process implies that specific mRNA molecules might be translationally regulated in response to eIF2α or S6 phosphorylation under these conditions.
mRNA translational engagement changes at late time points of DC-activation
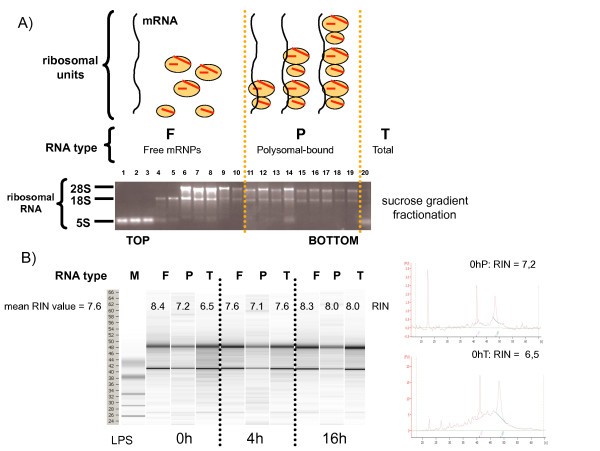
Efficient profiling and isolation of polysomes out of human moDCs. Polysomes sedimentation profiles (A) and RNA-integrity profiles (B) after sucrose gradient fractionation of untreated (0 h) or 4 h and 16 h LPS-stimulated moDCs.
In addition to non-activated cells (0 h), moDCs at 4 h and 16 h post-LPS stimulation were also chosen, since they represent two distinct activation states in which protein synthesis is either increasing (4 h), or decreasing (16 h), and in which the phosphorylation status of eIF2-α is radically different. No major difference in the polysomes sedimentation profiles was observed after the fractionation of the various DCs samples, confirming that actively translated mRNAs are present at all stages of DC maturation (not shown). To further identify translationally regulated mRNAs in maturing moDC, expression levels of all polysome-bound mRNAs were compared by microarray analysis (Affymetrix U133 Plus 2.0 GeneChip array comprising 54'675 probe sets) to those of unfractionated total mRNAs (translational profiling, Figure 2). Polysome-bound mRNAs (P) and total RNA (T) were isolated from moDCs generated from four different blood donors for three LPS activation time points (0 h, 4 h and 16 h), resulting in a total of twenty-four RNA samples to be analyzed. In addition to FACS characterization of the cells, the quality of the samples was further evaluated by comparing the total mRNA expression values of several DC-maturation markers from our experimental setting with available public databases values obtained under similar conditions [6]. Transcription of the co-stimulatory molecules CD80 and CD86, as well as the MHC I and II mRNAs were found to be all up-regulated after DC stimulation by LPS (see Additional file 1), thus confirming the quality of our samples and reliability of our analysis.
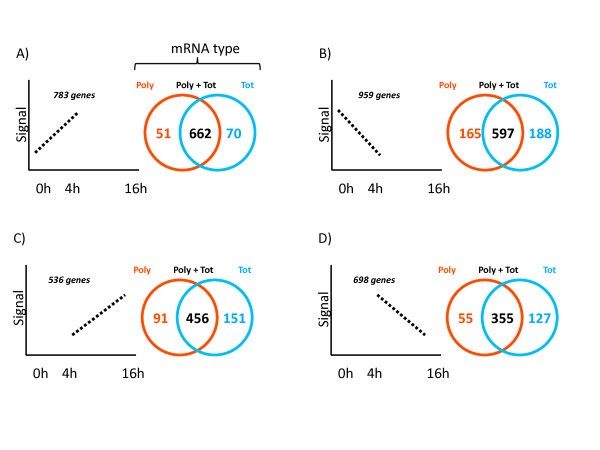
Global alterations of total and polysomal-bound mRNA in LPS-activated moDCs. The 54'675 probe sets present on the Affymetrix U133 Plus 2.0 GeneChip array were first filtered on expression (signal > 100 in all tested conditions) to obtain a preliminary list of 7'709 probe sets. The 7'709 probe sets were then filtered on fold change (applying a 2-fold cut-off) comparing Polysomal (Poly, red) and Total (Tot, blue) mRNA, between 0 h and 4 h (A and B) or 4 h and 16 h (C and D) post-LPS. For effective Venn diagram visualization, the transcriptionally up-regulated probe sets (A and C) were distinguished from the transcriptionally down-regulated probe sets (B and D). See Additional file 3 for a detailed description of the different genes subsets.
Biological functions of translationally regulated mRNAs in activated moDCs
We decided to take a statistically unbiased approach to further identify entire functional pathways, which could be regulated at the translational level. Importantly, the data obtained from four different donors were homogenous and no major pattern variation for both total and polysomal-bound RNA expression was found, thus allowing statistical analysis (see Additional file 3, compare the four columns within each T or P).
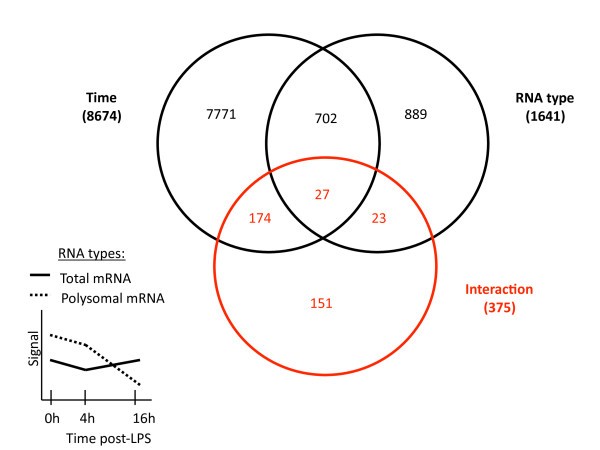
Statistical approach to identify translationally-regulated mRNA molecules in LPS-activated moDCs. The 54'675 probe sets present on the Affymetrix U133 Plus 2.0 GeneChip array were first filtered on flags (P in 50% 0 h or P in 50% 4 h or P in 50% 16 h) to obtain a preliminary list of 20'438 probe sets. A 2-way ANOVA analysis with repeats on time (false discovery rate= 0.05) was then performed on the 20'438 selected probe sets, to identify the 375 translationally-regulated mRNA molecules with statistically significant interaction (red circle in Venn diagram). An example of interaction (the two curves are parallel between 0 h and 4 h and are "interacting" between 4 h and 16 h post-LPS) between Total and Polysomal mRNA is indicated on the simplified graph on left, where the different time points of the two RNA groups are connected by their mean gene expression signals. The number of probe sets belonging to each parameter is indicated in brackets. See Additional file 4 for the complete list of translationally-regulated mRNA molecules. Groups defined within each parameter, Time: 0 h, 4 h and 16 h post-LPS; RNA type: Total mRNA and Polysomal mRNA.
Top "biological functions" of mRNA molecules affected by translation regulation in LPS-activated moDCs
Category (nr. of molecules) |
Biological Function |
p-value |
Molecules (gene symbols) |
|---|---|---|---|
Protein Synthesis (26) |
biosynthesis of proteins and translation regulation |
1.73E-04 |
ADAMTS5, CASP9, EEF2, EIF2A, EIF4A2, EIF4B, NCK1, PLAU, PSMC2, RBM3, RPL3, RPL5, RPL7, RPL13, RPL14, RPL15, RPL26, RPL31, RPL37, RPL38, RPL9, RPS11, SERPINB1, SRGN, UBC, UBE2K |
RNA Post-Transcriptional Modification (12) |
modification of mRNA |
8.67E-03 |
CDC5L, CPSF1, DBR1, EIF4A2, EIF4B, GRSF1, HNRNPR, CPSF1, GRSF1, MAPKAPK2, ZFP36, QARS |
Amino Acid Metabolism (11) |
catabolism of L-tryptophan and dephosphorylation of amino acids |
8.57E-08 |
CD80, IL6, INDO, KMO, KYNU, DUSP2, DUSP3, PPM1A, PPM1F, PPP1CB, PTPN6 |
Cell Morphology (10) |
transmembrane potential of mitochondria |
1.26E-03 |
AP2A2, BNIP3, CASP9, CFLAR, IL6, MAPK9, MOAP1, NFE2L2, PTPN6, SRGN |
Immune Cell Trafficking (8) |
emigration of leukocytes |
2.08E-03 |
CD44, CXCL3, CXCR4, CYTIP, GFI1, LDLR, PLAU |
Cancer (7) |
tumorigenesis of intestinal tissue |
2.64E-03 |
APC, MLH1, IFI16, SMARCA2, HSPA1A, IFI16, IGFBP4 |
Nucleic Acid Metabolism (5) |
metabolism of nucleic acid component |
6.15E-04 |
ATIC, MTAP, OAS1, OAS2, REXO2 |
Cell-mediated Immune Response (4) |
secretion of cytokine |
3.60E-03 |
CADM1, LCP2, SRGN, GFI1 |
Antigen Presentation (2) |
antigen peptide transporter |
8.84E-03 |
TAP1, TAP2 |
Small molecule biochemistry (1) |
activation by LPS |
4.21E-02 |
LY96 (MD2) |
The biological function with the most significative p-value for translational regulation was protein synthesis itself (26 molecules), including 3 translation factors among which was found eIF2α. Other identified pathways included "post-transcriptional modification" and "amino acid metabolism" comprising again some molecules involved in protein synthesis and amino acid modification (e.g. QARS, eIF4B, or INDO, DUSP2 and DUSP3).
As for molecules directly relevant to DC immune function, we identified genes involved in pathogen sensing (e.g.: OAS1, OAS2, LY96), antigen processing (e.g.: TAP1 and TAP2), immune regulation (e.g.: IL-6, INDO, CD80, SLP2) or leukocytes emigration (e.g. CXCL3 and CXCR4). Clearly this list indicates that a number of mRNAs expressed during DC maturation and important for their immuno-modulatory function are regulated at the translational level, such as Indoleamine 2,3-dioxygenase (IDO or INDO). IDO is a potent immuno-regulatory enzyme that degrades the essential amino acid tryptophan and results in a rise in uncharged tRNA, which activates the GCN2 kinase and downstream signaling such as the phosphorylation of eIF2α. Thus, IDO is likely to be preferentially translated in conditions of eIF2α phosphorylation and might therefore be regulated at the translational level.
Validation of the array data by real time PCR using total and polysome-bound RNA populations.
Gene Symbol |
Affy probe set |
RNA type |
Fold change (p-value) Array |
Fold change (+/- SD) qPCR |
||
|---|---|---|---|---|---|---|
4 h vs 0 h |
16 h vs 4 h |
4 h vs 0 h |
16 h vs 4 h |
|||
IL-6 |
205207_at |
Poly |
503,11 (0,040) |
-7,3 (0,101) |
3641,21 (11,92) |
-58,26 (1,21) |
Tot |
239,22 (0,010) |
-7,3 (0,051) |
2017,22 (19,13) |
-12,95 (2,11) |
||
RPL26 |
222229_x_at |
Poly |
1,41 (0,205) |
-1,78 (0,044) |
1,81 (0,32) |
-1,06 (0,33) |
Tot |
-1,36 (0,129) |
-1,33 (0,321) |
-3,12 (0,41) |
-1,52 (0,34) |
||
RPL14 |
213588_x_at |
Poly |
1,45 (0,008) |
-1,47 (0,101) |
0,31 (0,12) |
-0,81 (0,22) |
Tot |
-1,1 (0,191) |
-1,17 (0,089) |
-1,31 (0,32) |
-1,29 (0,33) |
||
RPS23 |
200926_at |
Poly |
1,73 (0,099) |
-2,74 (0,100) |
0,72 (0,13) |
-2,52 (0,35) |
Tot |
1,05 (0,011) |
-1,72 (0,111) |
0,32 (0,14) |
-1,89 (0,07) |
||
CD80 |
1554519_at |
Poly |
22,81 (0,133) |
1,31 (0,002) |
19,71 (0,72) |
0,77 (0,22) |
Tot |
33,65 (0,001) |
1,43 (0,005) |
22,71 (1,52) |
0,41 (0,11) |
||
OAS1 |
202869_at |
Poly |
11,56 (0,048) |
1,36 (0,044) |
7,26 (1,24) |
1,74 (0,71) |
Tot |
13,12 (0,026) |
1,86 (0.003) |
12,32 (0,32) |
1,26 (0,11) |
||
OAS2 |
204972_at |
Poly |
34,15 (0,043) |
-1,16 (0,111) |
7,51 (0.12) |
1,48 (0,45) |
Tot |
23,5 (0,001) |
1,2 (0,011) |
5,91 (0,11) |
1,21 (0,22) |
||
CASP9 |
203984_s_at |
Poly |
-3,94 (0,011) |
2,92 (0,009) |
-14,71 (1,3) |
2,79 (0,81) |
Tot |
-3,74 (0,005) |
2,02 (0,001) |
-11,7 (2,1) |
3,09 (0,62) |
||
HLA-F |
221875_x_at |
Poly |
2,07 (0,021) |
1,21 (0,008) |
3,41 (0,42) |
1,97 (0,32) |
Tot |
1,92 (0,002) |
1,97 (0,007) |
2,31 (0,71) |
2,25 (0,55) |
||
INDO |
210029_at |
Poly |
117,32 (0,022) |
1,34 (0,001) |
1176,21 (8,33) |
2,01 (0,26) |
Tot |
97,51 (0,011) |
1,91 (0,008) |
737,2 (3,55) |
2,54 (0,27) |
||
TAP1 |
202307_s_at |
Poly |
6,62 (0,340) |
1,02 (0,009) |
4,71 (1,1) |
0,79 (0,20) |
Tot |
8,38 (0,013) |
1,31 (0,002) |
6,2 (0,89) |
1,55 (0,41) |
||
TAP2 |
225973_at |
Poly |
3,19 (0,001) |
1,56 (0,111) |
3,01 (0,66) |
1,62 (0,61) |
Tot |
3,20 (0,013) |
1,95 (0,505) |
3,21 (0,52) |
1,93 (1,10) |
||
MD2 |
206584_at |
Poly |
-1,37 (0,008) |
-3,43 (0,043) |
-2,91 (0,77) |
-3,30 (0,21) |
Tot |
1,11 (0,008) |
-2,31 (0,002) |
1,51 (0,33) |
-2,10 (0,41) |
||
eIF4B |
211938_at |
Poly |
-2,26 (0,001) |
-1,65 (0,031) |
-2,21 (0,23) |
0,87 (0,04) |
Tot |
-3,81 (0,006) |
-1,66 (0,003) |
-17,91 (2,21) |
0,35 (0,02) |
||
Specific ribosomal proteins are translationally regulated in activated moDCs
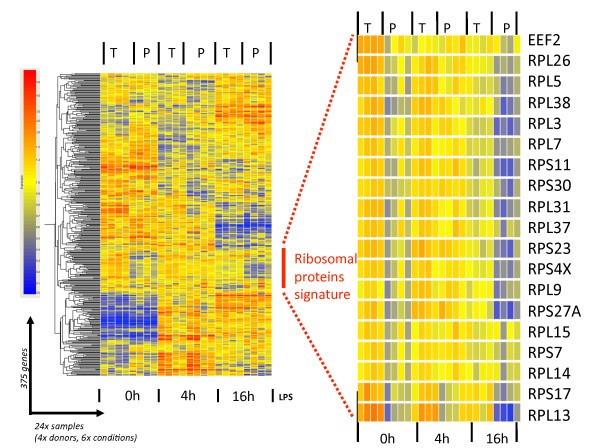
Specific ribosomal protein mRNAs are translationally-regulated in LPS-activated moDCs. The 375 mRNA molecules with statistically significant interaction (see Figure 4) were clustered on a heat-map using the software GeneSpring GX 7.3. The translationally regulated ribosomal protein mRNAs appear as a specific signature (left panel), which has been extracted and enlarged (right panel). See the text for more details.
In recent years, several reports have also suggested that translation of TOP mRNAs can be independent of S6 phosphorylation [20, 21]. In activated DCs, S6 phosphorylation correlates well with the translational engagement of the ribosomal proteins after 4 h of LPS stimulation (Figure 5, 4h). However even if the intensity of S6 phosphorylation keeps increasing during the following 12 h of DC maturation, several of the large ribosomal protein mRNAs are nevertheless clearly disengaged from polysomes. Thus, although S6 phosphorylation is likely to favor translational engagement of 5'-TOP containing mRNAs, it does not counteract their increased release from the ribosomes.
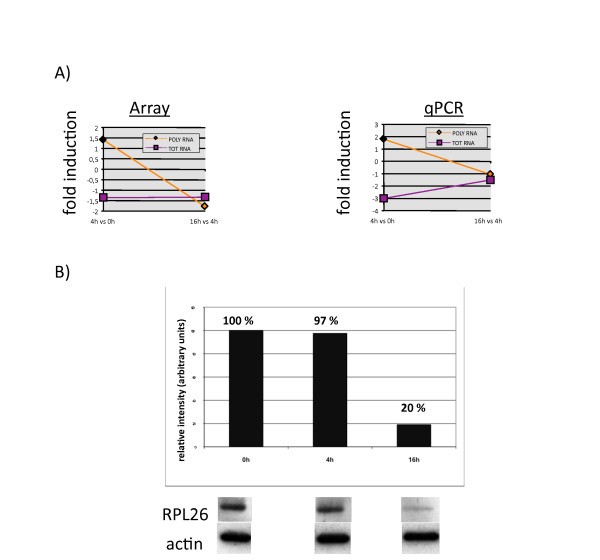
Correlation between RPL26 mRNA translational disengagement and protein down-regulation in LPS-activated moDCs. (A) Gene expression of the RPL26 Total and Polysomal mRNAs determined by microarrays analysis (left) and confirmed by qRT-PCR analysis (right), depicted as fold induction between 4 h and 0 h and 16 h and 4 h post-LPS. (B) Immunoblot to assay RPL26 protein expression at 0 h, 4 h and 16 h post-LPS. An actin immunoblot is shown for equal loading control. The relative protein expression (top) has been determined by quantifying the immublot signals with the software ImageQuant (Fuji) and is representative of a typical experiment (n = 3).
The translation regulation of ribosomal protein mRNAs is one of the features of LPS-activated human DCs. In the early phase of DC maturation, a specific increase in ribososomal proteins synthesis might impact positively on the global translation enhancement in response to TLR-4 engagement and subsequent PI3K activation, while a reduction at later time might favor a decrease in translation intensity. The reduction in RPL26 might also have a specific effect on the survival of activated DCs. Although many aspects of the non-translational role of ribosomal proteins remain to be investigated, it is noteworthy to underline that several ribosomal protein (RLP4, RLP22 and RLP35) and translation factors (eEF1d) mRNAs represent the most significant genes up-regulated in a statistical group comparison performed to identified genes differentially expressed in Systemic onset juvenile idiopathic arthritis (SoJIA) patients compared with healthy children [24]. SoJIA patients are also characterized by abundant production of interleukin (IL)-1, which is an important mediator of this disease and can be induced in DCs by LPS, thus creating an additional link between 5'-TOP containing mRNAs and inflammatory conditions. Our results on the characterization of RPL26 mRNA and its protein expression strongly support our conclusion that other RLP genes belong to a cluster being translationally regulated during DC maturation. Translation regulation therefore appears as a key function to control specific gene expression and more precise analysis combining traditional gene expression arrays and translation profiling will have to be carried-out to follow the immune response and host-pathogen interactions and single-out functionally important mRNAs regulated at the translation level.
Methods
Cell Culture
Fresh human leukapheresis products were obtained from the EFS (Marseille, France). Human peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-PaqueTM PLUS (Amersham Biosciences, Uppsala, Sweden), washed four times with RPMI and CD14+ cells were immunomagnetically purified with AutoMACS system following the protocol of the manufacturer (Miltenyi Biotech, Auburn, CA). Purified CD14+ monocytes were analyzed using a FACSCalibur (Becton Dickinson, San Jose, CA), confirming the purity of CD14+ cells to be 95%. To promote differentiation into iDC, the purified CD14+ cells (0.5 × 106 cells/ml) were plated in 6-well plates (2 × 106 cells/well) and cultured in RPMI 1640 medium supplemented with 10% FCS, non essential amino acids, penicillin/streptomycin 100 ng/mL (>1000 U/ml), recombinant human GM-CSF and 20 ng/mL (>100 U/ml) IL-4 for 5 days (both from PeproTech, Rocky Hill, NJ). At days 2 and 4, half of the volume of the medium was replaced by fresh medium supplemented with GM-CSF and IL-4. For DC maturation, 100 ng/mL LPS (Escherichia coli type 026:B6; Sigma-Aldrich, St. Louis, MO) was added to the cells at day 5, for the indicated number of hours.
Polysomal profiling by sucrose gradient fractionation
Polysome-bound mRNA molecules were enriched by sucrose gradient fractionation following the protocol originally developed by Garcia-Sanz and collaborators [25]. Briefly, 60 to 80 × 106 day 5 human moDCs were lysed in 1 ml of polysome buffer (10 mM Tris-HCl (pH 8), 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP40, 0.1 mg/ml cycloheximide, and 500 units/mL RNasin (Promega, Madison, WI). After 10 min on ice, lysates were quickly centrifuged (10.000 × g for 10 sec at 4°C) and the supernatant was resuspended in a stabilizing solution (0.2 mg/ml cycloheximide, 0.7 mg/ml heparin, 1 mM phenylmethanesulfonyl fluoride). After a quick centrifugation (12.000 × g for 2 min at 4°C) to remove mitochondria and membrane debris, the resulting supernatant was layered on a 15% to 40% sucrose gradient. Gradients were then ultracentrifuged (35.000 × g for 2 h at 4°C, SW41 rotor) and after centrifugation 20 × 550 ml fractions were collected, starting from the top of the gradient. All the fractions were then digested with Proteinase K (200 mg/ml) in presence of 1%SDS and 10 mM EDTA. RNA was then extracted with Phenol/Chloroform/Isoamylalcohol (volume ratio 25:24:1) and precipitated with 2.5 Volumes of 100% Ethanol in presence of 0.8 M lithium chloride, necessary to get rid of heparin, a known inhibitor of RT activity [26]. After precipitation all the RNA were resuspended in 20 ml RNase free H2O. The correct fractionation of the polysomes was tested by detecting the different rRNA types on a 1% denaturing agarose gel. Total RNA was directly extracted out of moDCs without fractionation. Total and polysomal-bound RNAwere purified using the RNeasy miniprep kit (Qiagen, Chatsworth, CA). To exclude theamplification of genomic DNA, an on-column DNase digestion was performed using the RNase-Free DNase Set (Qiagen). The RNA Integrity Number (RIN-value) of all RNA types and timepoints was measured with the Agilent 2100 Bioanalyzer. RIN-values between 6.5 and 8.5 (mean RIN value = 7.6) were obtained, indicating that the RNA had sufficient integrity to be analyzed by microarrays.
Affymetrix microarray hybridization and data mining
For each condition 100 ng of total or polysomal-bound RNA were employed to synthesize double-stranded cDNA using two successive reverse-transcription reactions according to standard Affymetrix protocols (GeneChip Two-Cycle Target Labelling, Affymetrix, Santa Clara, CA). Linear amplification with T7-RNA polymerase and biotin labelling were performed by in vitro transcription by standard Affymetrix procedures. The resulting biotin-labeled cRNA was fragmented and hybridized to the Affymetrix Human Genome U133 2.0 oligonucleotide 14,500-gene microarray chip for 16 h at 45°C. Following hybridization, the probe array was washed and stained on a fluidics station and immediately scanned on a Affymetrix GCS 3000 GeneArray Scanner. The data generated from the scan were then analyzed using the MicroArray Suite software (MAS 5.0, Affymetrix). The data derived from four independent experiments were normalized using the GC-RMA algorithm and bioinformatic analysis was performed using GeneSpring GX 7.3 (Agilent, Palo Alto, CA) and Statistics Analysis System (SAS v9.1.3). Probe selection was performed using 2-way ANOVAs accounting for repeated measures with a false discovery rate of 0.05. Hierarchical clustering was performed using the default clustering algorithm and setting in GX7.3.
Quantitative real-time RT-PCR
Total RNA was extracted and purified using the RNeasy kit (Qiagen). To exclude the amplification of genomic DNA, an on-column DNase digestion was performed using the RNase-Free DNase Set (Qiagen). 1 μg of RNA was retro-transcribed using SuperScript II reverse transcriptase (Invitrogen) and random (pDN6) primers. First-strand cDNA templates were then used for PCR amplification of short (100 to 150 bp) exon fragments of the gene of interest using the appropriate primers (Additional file 4 shows the complete list of the 375 probe sets with statistically significant interaction). PCR was carried out using a Stratagene MX3000P Real-Time PCR System in complete SYBR Green PCR buffer (PE Biosystems, Warrington, UK) using 200 nM of each specific primer. A total of 20 μl of PCR mix was added to 5 μl of cDNA template, and the amplification was tracked via SYBR Green incorporation by using a Stratagene (La Jolla, CA) sequence detection system 4.01. Comparative real-time PCR was done in triplicate, including no-template controls. A dissociation curve was generated at the end of each PCR cycle to verify that a single product was amplified. Relative quantification of target cDNA was determined by calculating the difference in cross-threshold (C t) values after normalization to GAPDH signals, according to the Pfaffl method and the automated Excel-based program available (REST©). The sequences of all employed primers are available in Additional file 7.
Immunodetection and antibodies
50 μg of TX-100 soluble material or 200.000 cells, lysed directly in Laemmli buffer, were loaded on 10% SDS-PAGE prior to immunoblotting and chemiluminescence detection (SuperSignal, Pierce, USA). SUnSET was performed as previously described [14] using A647-labelled mouse IgG2a anti-puromycin antibody (12D10). Antibody against RPL26 was from Abnova. For flow cytometry analysis, APC-conjugated anti-HLA-DR (L243 clone) and PE-conjugated anti-CD86 (clone IT2.2) were from BD PharMingen (San Jose, CA, USA). Anti- eIF2, phospho-eIF2 and -phospho-S6 were from Cell Signaling Technology (Beverly, MA, USA). For immunofluorescence analysis, MoDCs were let adhere on Alcyan blue coated-coverslips and surface stained with unlabelled antibody at 4°C for 30 min. After washes, coverslips were placed in warm medium for the indicated time and then fixed in 3% PFA for conventional staining with secondary antibodies.
GEO Reviewers Link
Following link has been created to allow read-only access review of record GSE14000 and associated accessions, which are the Affymetrix gene expression data related to the submitted manuscript.
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=jlalxuycqigwqnw&acc=GSE14000
Declarations
Acknowledgements
This work is supported by grants to PP from the ANR- DC-Trans, Réseau National Génopole, La Ligue Nationale Contre le Cancer and the Human Frontier of Science Program. MC is supported by the Swiss National Science Foundation (SNF). PP is part of the DC-THERA FP6 NoE. We are particularly indebted to the CNRS Affimetrix facility and to the PICsL imaging core facility for expert technical assistance.
Authors’ Affiliations
References
- Steinman RM: Lasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune system. Nat Med 2007, 13:1155–1159.View ArticlePubMedGoogle Scholar
- Lelouard H, Schmidt EK, Camosseto V, Clavarino G, Ceppi M, Hsu HT, Pierre P: Regulation of translation is required for dendritic cell function and survival during activation. J Cell Biol 2007, 179:1427–1439.View ArticlePubMedGoogle Scholar
- Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM: Activation of the integrated stress response during T helper cell differentiation. Nat Immunol 2006, 7:644–651.View ArticlePubMedGoogle Scholar
- Sonenberg N, Hinnebusch AG: New modes of translational control in development, behavior, and disease. Mol Cell 2007, 28:721–729.View ArticlePubMedGoogle Scholar
- Pierre P: Immunity and the regulation of protein synthesis: surprising connections. Curr Opin Immunol 2009, 21:70–77.View ArticlePubMedGoogle Scholar
- Huang Q, Liu do N, Majewski P, Schulte le AC, Korn JM, Young RA, Lander ES, Hacohen N: The plasticity of dendritic cell responses to pathogens and their components. Science 2001, 294:870–875.View ArticlePubMedGoogle Scholar
- Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, Xu H, Vivier E, Sellars M, Pierre P, et al.: Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol 2008, 9:R17.View ArticlePubMedGoogle Scholar
- Todd DJ, Lee AH, Glimcher LH: The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 2008, 8:663–674.View ArticlePubMedGoogle Scholar
- Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P: MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci USA 2009, 106:2735–2740.View ArticlePubMedGoogle Scholar
- Pradet-Balade B, Boulme F, Beug H, Mullner EW, Garcia-Sanz JA: Translation control: bridging the gap between genomics and proteomics? Trends Biochem Sci 2001, 26:225–229.View ArticlePubMedGoogle Scholar
- Beilharz TH, Preiss T: Translational profiling: the genome-wide measure of the nascent proteome. Brief Funct Genomic Proteomic 2004, 3:103–111.View ArticlePubMedGoogle Scholar
- Parent R, Beretta L: Translational control plays a prominent role in the hepatocytic differentiation of HepaRG liver progenitor cells. Genome Biol 2008, 9:R19.View ArticlePubMedGoogle Scholar
- Warner JR, McIntosh KB: How common are extraribosomal functions of ribosomal proteins? Mol Cell 2009, 34:3–11.View ArticlePubMedGoogle Scholar
- Schmidt EK, Clavarino G, Ceppi M, Pierre P: SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 2009, 6:275–277.View ArticlePubMedGoogle Scholar
- Ron D, Walter P: Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007, 8:519–529.View ArticlePubMedGoogle Scholar
- Sabatini DM: mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006, 6:729–734.View ArticlePubMedGoogle Scholar
- Kiewe P, Gueller S, Komor M, Stroux A, Thiel E, Hofmann WK: Prediction of qualitative outcome of oligonucleotide microarray hybridization by measurement of RNA integrity using the 2100 Bioanalyzer capillary electrophoresis system. Ann Hematol 2009, 88:1177–1183.View ArticlePubMedGoogle Scholar
- Levy S, Avni D, Hariharan N, Perry RP, Meyuhas O: Oligopyrimidine tract at the 5' end of mammalian ribosomal protein mRNAs is required for their translational control. Proc Natl Acad Sci USA 1991, 88:3319–3323.View ArticlePubMedGoogle Scholar
- Meyuhas O: Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 2000, 267:6321–6330.View ArticlePubMedGoogle Scholar
- Stolovich M, Tang H, Hornstein E, Levy G, Cohen R, Bae SS, Birnbaum MJ, Meyuhas O: Transduction of growth or mitogenic signals into translational activation of TOP mRNAs is fully reliant on the phosphatidylinositol 3-kinase-mediated pathway but requires neither S6K1 nor rpS6 phosphorylation. Mol Cell Biol 2002, 22:8101–8113.View ArticlePubMedGoogle Scholar
- Ruvinsky I, Meyuhas O: Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 2006, 31:342–348.View ArticlePubMedGoogle Scholar
- Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M: Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell 2008, 32:180–189.View ArticlePubMedGoogle Scholar
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H: Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 2004, 24:7654–7668.View ArticlePubMedGoogle Scholar
- Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, Ardura M, Chung W, Wise C, Palucka K, et al.: Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med 2007, 204:2131–2144.View ArticlePubMedGoogle Scholar
- Garcia-Sanz JA, Mikulits W, Livingstone A, Lefkovits I, Mullner EW: Translational control: a general mechanism for gene regulation during T cell activation. FASEB J 1998, 12:299–306.PubMedGoogle Scholar
- del Prete MJ, Vernal R, Dolznig H, Mullner EW, Garcia-Sanz JA: Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. Rna 2007, 13:414–421.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.