
PREDTAP: a system for prediction of peptide binding to the human transporter associated with antigen processing
- Software
- Open Access
PREDTAP: a system for prediction of peptide binding to the human transporter associated with antigen processing
- Received: 14 January 2006
- Accepted: 23 May 2006
- Published: 23 May 2006
Abstract
Background
The transporter associated with antigen processing (TAP) is a critical component of the major histocompatibility complex (MHC) class I antigen processing and presentation pathway. TAP transports antigenic peptides into the endoplasmic reticulum where it loads them into the binding groove of MHC class I molecules. Because peptides must first be transported by TAP in order to be presented on MHC class I, TAP binding preferences should impact significantly on T-cell epitope selection.
Description
PREDTAP is a computational system that predicts peptide binding to human TAP. It uses artificial neural networks and hidden Markov models as predictive engines. Extensive testing was performed to valid the prediction models. The results showed that PREDTAP was both sensitive and specific and had good predictive ability (area under the receiver operating characteristic curve Aroc>0.85).
Conclusion
PREDTAP can be integrated with prediction systems for MHC class I binding peptides for improved performance of in silico prediction of T-cell epitopes. PREDTAP is available for public use at [1].
Keywords
- Major Histocompatibility Complex
- Artificial Neural Network
- Hide Markov Model
- Major Histocompatibility Complex Class
- Artificial Neural Network Model
Background
Peptides that bind major histocompatibility complex (MHC) class I molecules serve as recognition targets for cytotoxic CD8+ T cells (CTLs). The major function of CTLs is recognition and destruction of infected (e.g. viruses, bacteria, parasites or fungi), mutated (e.g. cancer), or foreign (e.g. transplants) cells. CTLs recognize short antigenic peptides (T-cell epitopes) presented by MHC class I molecules that mainly originate from degradation of cytosolic proteins. Intracellular antigen processing pathways determine the selectivity of peptides which are available for binding to MHC class I molecules and are thereby important targets of CTL responses [2].
MHC class I antigen processing pathway steps include proteosomal cleavage of proteins into shorter peptides, translocation of peptides into the endoplasmic reticulum (ER) by TAP, optional ER trimming by aminopeptidases, insertion of peptides into the binding groove of MHC molecules, and transport of peptide/MHC complexes to the cell surface for presentation to CTLs [3]. TAP is a transmembrane protein responsible for the transport of antigenic peptides into the ER. TAP demonstrates peptide binding selectivity and the affinity of a particular peptide for TAP influences the probability of its presentation by MHC class I molecules. Peptides that are 8–16 amino acids long and have sufficient binding affinity are efficiently translocated by TAP into the ER, while longer peptides may be transported but with lower efficiency [4]. Human TAP (hTAP) is a heterodimer that has two subunits hTAP1 and hTAP2. TAP belongs to the ATP-binding cassette transporters and each subunit protein has one transmembrane domain and one ATP-binding binding domain. The genes for human TAP1 and TAP2 are located in the MHC II locus of chromosome 6 and comprise 10 kb each [5]. A more detailed description of function, structure, expression of TAP can be found in [6].
The efficiency of TAP-mediated translocation of a peptide is proportional to its TAP-binding affinity [7, 8]. Mutations, such as premature stop codons, or deletions of either hTAP1 or hTAP2 impair peptide transport into ER and result in a significant reduction of surface expression of peptide/MHC complexes [9]. TAP deficient cells have low cell-surface HLA class I expression shown to range from 10% (HLA-A2) to 3%, (HLA-B27 and -A3) [10]. The majority of the peptides presented by HLA class I on cell surface are thus dependent on TAP.
Identification of T-cell epitopes is a highly combinatorial problem. The diversity of human immune responses to T-cell epitopes originates from two sources – high allelic variation of the host (both HLA molecules and T-cell receptors) and high variation of target antigens, particularly those derived from viruses. Computational models are routinely used for pre-screening of potential T-cell epitopes and minimization of the number of necessary experiments. Most developments have focused on modeling and prediction of peptide binding to MHC molecules [see [11]]. Amongst computational models of peptide binding to hTAP that have been developed are binding motifs [7], quantitative matrices [12, 13, 14], artificial neural networks (ANN) [12, 15], and support vector machines (SVM) [16]. Combined computational methods that integrate multiple critical steps – proteasome cleavage, TAP transport, and MHC class I binding have been proposed as a supporting methodology for prediction of high probability targets for therapeutic peptides and vaccines [17]. Several combined computational applications of models of antigen processing and presentation have been reported [18, 19, 20, 21, 22]. Testing results indicate that these predictions produce a lower incidence of false positives and reduce the number of experiments required for identification of T-cell epitopes. However, these combined predictions need to be taken with a dose of caution. Alternative pathways for both proteolytic degradation [23] and TAP transport [24] have been reported. In some cases TAP-deficient individuals have normal immune responses [25], suggesting that TAP-independent immune responses are sufficient to provide effective protection from some intracellular pathogens. Nevertheless, the proteasome-TAP-MHC class I pathway is responsible for 90–97% of expression of peptide/MHC Class I complexes and therefore is critical for the identification of target epitopes for immunotherapies and vaccines.
We developed PREDTAP, a computational system that predicts peptides binding to hTAP. It uses ANN and hidden Markov models (HMM) as predictive engines. Extensive testing was performed to validate the prediction models and ensure that PREDTAP is both sensitive and specific. PREDTAP is available for public use at [1].
Materials and methods
Training dataset
Number of peptides in the training dataset
Binding Affinity |
Number of peptides |
|---|---|
0 |
26 |
1 |
52 |
2 |
48 |
3 |
48 |
4 |
53 |
5 |
55 |
6 |
40 |
7 |
87 |
8 |
61 |
9 |
16 |
10 |
7 |
Sum |
493 |
Artificial Neural Network
3-layer backpropagation ANN models (in-house software) were used for the development of the PREDTAP server. The learning method was error backpropagation with a sigmoid activation function. The inputs to the ANN were the binary strings representing nonamer peptides. There are twenty naturally-occurring amino acids encoded by the standard genetic code. Each amino acid in a nonamer peptide can be encoded as a binary string of length 20 with a unique position set to "1" and other positions set to "0", resulting in a binary string of length 180 to represent the nonamer. For example the first two amino acids, by alphabetic order, alanine (A) and cysteine (C) are encoded by 10000000000000000000 and 01000000000000000000 respectively, and the last amino acid tyrosine (Y) is encoded by 00000000000000000001. The outputs were binding scores ranging from zero to ten. The higher the score, the higher the possibility of the peptide being a TAP binder. Two ANN architectures were used, 180-2-1 and 180-1-1. The maximum number of the ANN training cycles was set to 300. The training was repeated for four times, and four sets of weights were obtained. The value of momentum was 0.5 and of learning rate 0.2. The error threshold for stopping training was 0.01.
Hidden Markov Model
HMMs have been applied successfully in prediction of HLA class I-binding peptides [26, 27]. An HMM is defined by a finite set of states representing possible states of the modeled system. Some of these states may be directly observable, but some are not, and are denoted as hidden. Biological problems are often sequential and HMM frequently utilize sequential ordering of system states. A change (transition) of the system from one state to another is governed by statistical regularities. The probability distribution of the system states can be estimated from the data. In the present study, we used a first-order HMM, in which the current system state is determined only by the preceding state, as described in [26].
Cross-validation
Cross-validation is a method for error rate estimation. It implements a simple idea: the dataset of size n samples is partitioned into two parts, the model parameters are estimated using one set and the goodness-of-fit criterion evaluated on the second set. The cross-validation estimates the goodness-of-fit criterion. Cross-validation tends to overfit when selecting a correct model – it may choos an overly-complex model for the given dataset. There is some evidence that for model selection multifold cross-validation, where more than one samples are deleted form the training set in each comparison, performs better than a simple leave-one-out cross-validation[28]. In our experiments, 10-fold cross-validation was performed to evaluate the performance of the classifiers.
Prediction performance measurement
The predictive performance of the models was evaluated by sensitivity (SE) and specificity (SP) measures. Sensitivity, SE = TP/(TP+FN), indicates percentage of correctly predicted binders, where TP stands for number of true positive predictions (experimental binder predicted as binder) and FN stands for number of false negative predictions (experimental binder predicted as non-binder). Specificity, SP = TN/(TN+FP), indicates percentage of correctly predicted non-binders, where TN stands for number of true negative predictions (experimental non-binder predicted as binder) and FP stands for number of false positive predictions (experimental non-binder predicted as binder). For the studied problem, we consider values of SP >0.8 useful in practice.
The receiver operating characteristic (ROC) curve analysis provided a measure for overall prediction accuracies of prediction models [29]. The ROC curve is generated by plotting SE against (1-SP) for various classification thresholds. As a rough guide, the area under ROC (Aroc) value 1.0 represents a perfect prediction, values 0.9 to 1.0 represent excellent accuracy, 0.8 to 0.9 represent good accuracy, 0.7 to 0.8 represent marginal accuracy, 0.5 to 0.7 represents poor accuracy, while 0.5 represent predictions that indicate random choice [29].
The prediction performance of PREDTAP(ANN & HMM) was compared with that of publicly available predictive systems, TAPPred (SVM & cascade SVM) [16] and SVMTAP [19]. Three proteins, human papillomavirus type 16 E6 (P03126) with experimentally identified HLA-A3 binders [30], E7 (P03129) with a single HLA-A3 binder [30] peptides and human cancer antigen KM-HN-1 (NP_689988.1) with three HLA-A24 restricted T-cell epitopes [31], were used and the predicted TAP binders were compared with the HLA binding peptides.
Normalization of prediction scores
Brusic et al. [15] showed that ANN models were skewed with a tendency to center-shift prediction of both very low and very high TAP binders. To obtain prediction scores evenly distributed in the range 0–10, we have implemented prediction score normalization. The raw prediction scores produced by HMM methods are not within the range 0–10. Score mapping is also necessary to bring final prediction scores within the range 0–10. The mapping of scores was done according to equation:
scoren = (score - scoremin) / (scoremax - scoremin) × 10
score n denotes the normalized score, score denotes the raw prediction score, score min and score max denote the minimum and maximum values of the raw scores. The values for score min and score max were obtained using extensive simulation. More than 5000 randomly selected nonamer peptides were used for prediction using the ANN/HMM models. Since the testing data contains large number of nonamer peptides, the highest and lowest predicted score from the testing data were taken as reasonable maximum and minimum scores for normalization.
Implementation
The web interface of PREDTAP uses a set of Graphical User Interface forms. The interface was built using a combination of Perl, CGI and C programs. PREDTAP has been implemented in the SunOS 5.9 UNIX environment.
Model validation
Performance assessment of ANN/HMM models using 10-fold cross-validation
ANN 180-2-1 |
H |
MH |
LMH |
|---|---|---|---|
1st run |
0.95 |
0.95 |
0.89 |
2nd run |
0.95 |
0.94 |
0.88 |
3rd run |
0.95 |
0.94 |
0.88 |
ANN 180-1-1 |
H |
MH |
LMH |
|---|---|---|---|
1st run |
0.93 |
0.94 |
0.87 |
2nd run |
0.92 |
0.92 |
0.86 |
3rd run |
0.93 |
0.94 |
0.88 |
HMM |
H |
M |
L |
|---|---|---|---|
1st run |
0.9 |
0.9 |
0.87 |
2nd run |
0.89 |
0.9 |
0.87 |
3rd run |
0.89 |
0.9 |
0.86 |
The specificity vs. sensitivity plot of the ANN prediction model for prediction can be viewed at supplementary materials A [1]. The specificity/sensitivity plot of the HMM prediction model can be viewed at supplementary materials B [1].
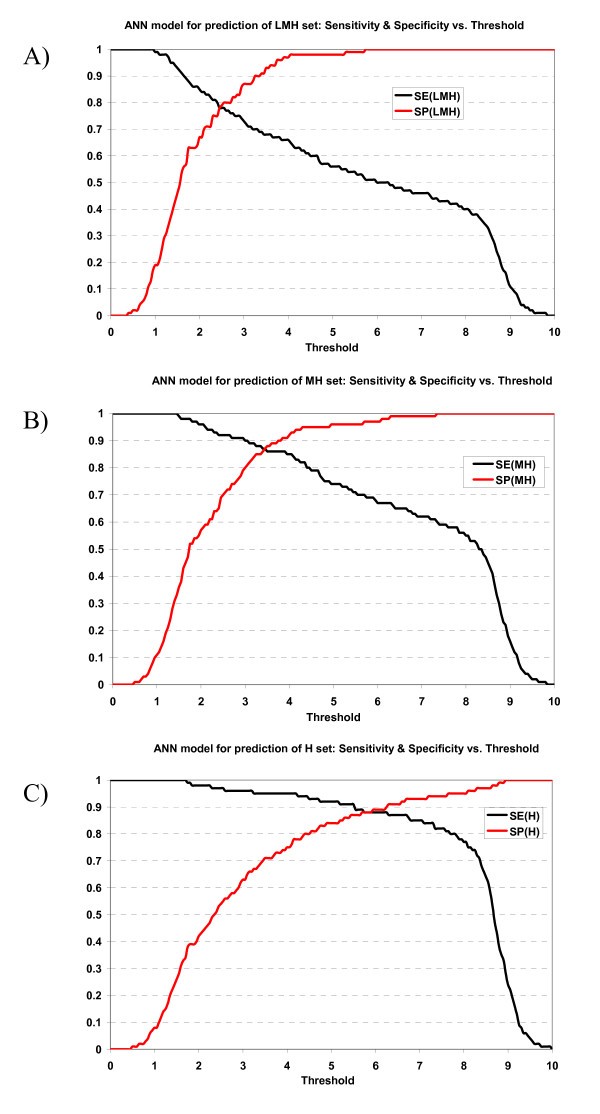
Plot of sensitivity and specificity of ANN model against thresholds in 10-fold cross-validation. The ANN model for prediction of A) LMH set, B) MH set, and C) H set.
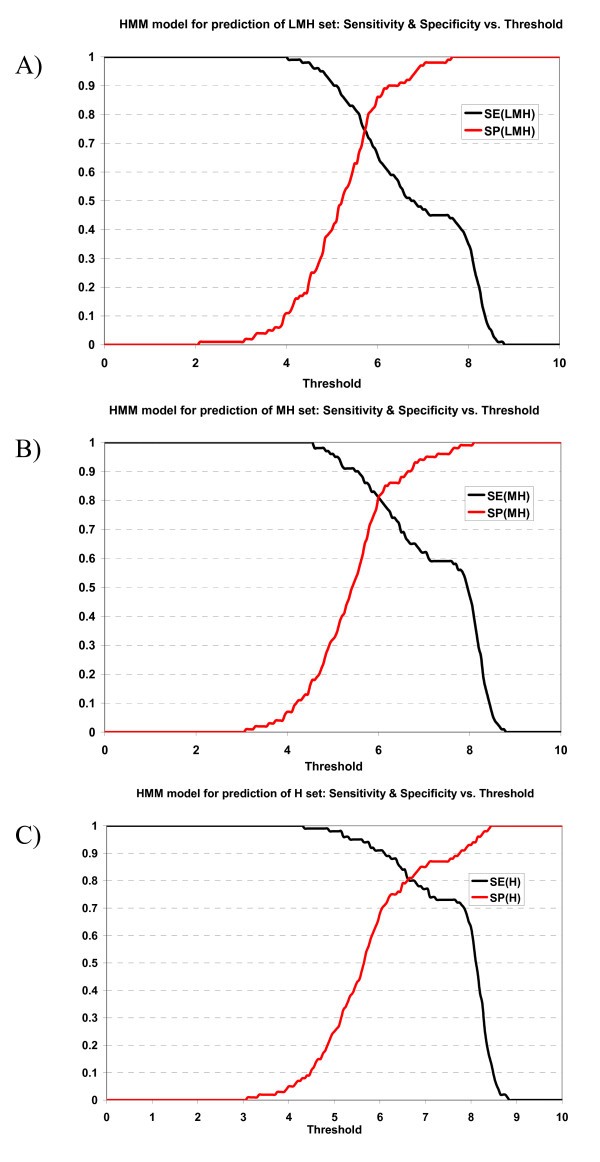
Plot of sensitivity and specificity of HMM model against thresholds in 10-fold cross-validation. The HMM model for prediction of A) LMH set, B) MH set, and C) H set.
Sensitivities and specificities of ANN and HMM models at the selection threshold 6.0
Threshold |
ANN |
SE |
SP |
|---|---|---|---|
6.0 |
LMH |
0.50 |
1.00 |
MH |
0.67 |
0.97 |
|
H |
0.88 |
0.89 |
Threshold |
HMM |
SE |
SP |
|---|---|---|---|
6.0 |
LMH |
0.66 |
.86 |
MH |
0.81 |
0.81 |
|
H |
0.91 |
0.68 |
Performance assessment of ANN/HMM models when the dataset was partitioned into two parts with the training dataset containing two thirds of the data points randomly selected and the testing set containing the remaining one third of data points
ANN 180-2-1 |
H |
MH |
LMH |
|---|---|---|---|
1st run |
0.91 |
0.92 |
0.85 |
2nd run |
0.96 |
0.95 |
0.90 |
3rd run |
0.94 |
0.91 |
0.87 |
HMM |
H |
M |
L |
|---|---|---|---|
1st run |
0.88 |
0.88 |
0.86 |
2nd run |
0.86 |
0.88 |
0.83 |
3rd run |
0.91 |
0.9 |
0.82 |
Comparison to other predictive systems
Amino acid position of top 5% predicted TAP binders in Human papillomavirus type 16 E6 (P03126) by SVMTAP, TAPPred and PREDTAP. The positions marked by "+" were selected by four prediction models. The positions marked by "*"were selected by three prediction models. The experimentally identified HLA-A*0301 binders are 17–15, 233–41, 342–50, 459–67, 575–83, 689–97, 793–101, and 8125–133). The predictions in the table marked by 1–8 are within 16-mers containing respective HLA-A*0301 binders
SVMTAP |
TAPPred (SVM) |
TAPPred (Cascade SVM) |
PREDTAP (ANN) |
PREDTAP (HMM) |
|---|---|---|---|---|
755,+ |
534,+ |
51 |
755,+ |
755,+ |
1318 |
68 |
604 |
534,+ |
463,* |
534,+ |
805 |
493 |
463,* |
614 |
150 |
815 |
1328 |
68 |
473 |
1308 |
755,+ |
937 |
83 |
71 |
463,* |
1318 |
116 |
594 |
534,+ |
134 |
134 |
67 |
423 |
493 |
146 |
51 |
402,3 |
1308 |
836 |
Amino acid position of the top 5% predicted TAP binders in HPV 16 E7 (P03129) by SVMTAP, TAPPred and PREDTAP. The positions marked by "+" were selected by four prediction models and those marked by "*"were selected by three prediction models. The experimentally identified HLA-A*0201 binder is 89–97. 1Within a 16-mer containing E7 89–97
SVMTAP |
TAPPred (SVM) |
TAPPred (Cascade SVM) |
PREDTAP (ANN) |
PREDTAP (HMM) |
|---|---|---|---|---|
49+ |
49+ |
58 |
50* |
49+ |
9* |
50* |
57 |
9* |
44 |
50* |
17 |
881 |
49+ |
43 |
59 |
9* |
821 |
48 |
71 |
7 |
59 |
67 |
76 |
3 |
Amino acid position of top 3% predicted TAP binders in the tumor antigen KM-HN-1 (NP_689988.1) by SVMTAP, TAPPred and PREDTAP. The positions marked by "+" were selected by four prediction models and those marked by "*"were selected by three prediction models. The predicted TAP-binders in proximity of known T-cell epitopes are designated by 1(196–204), 2(499–508) and 3(770–778)
SVMTAP |
TAPPred (SVM) |
TAPPred (Cascade SVM) |
PREDTAP (ANN) |
PREDTAP (HMM) |
||
|---|---|---|---|---|---|---|
Position |
Position |
Position |
Position |
Score |
Position |
Score |
660+ |
372+ |
674 |
1951,+ |
8.15 |
682 |
7.01 |
372+ |
1951,+ |
314 |
654* |
8.09 |
5062,+ |
6.98 |
426* |
426* |
639 |
372+ |
8.06 |
372+ |
6.87 |
1951,+ |
794 |
249 |
422 |
7.94 |
683 |
6.83 |
794 |
330 |
530 |
565 |
7.41 |
507+ |
6.65 |
199 |
317+ |
525 |
317+ |
6.53 |
1951,+ |
6.62 |
654* |
660+ |
325 |
310 |
5.97 |
492 |
6.44 |
317+ |
331 |
206 |
378 |
5.59 |
310 |
6.41 |
371 |
652* |
12 |
468 |
5.54 |
660+ |
6.41 |
110 |
199 |
479 |
7633 |
5.48 |
16 |
6.37 |
760 |
198 |
537 |
337 |
5.36 |
468 |
6.33 |
198 |
371 |
112 |
426* |
5.33 |
317+ |
6.19 |
789 |
654* |
93 |
737 |
5.16 |
395* |
6.16 |
705 |
5062,+ |
626 |
246 |
5.04 |
573 |
6.12 |
457 |
789 |
470 |
660+ |
4.98 |
223 |
6.12 |
507+ |
457 |
141 |
756 |
4.96 |
193 |
6.09 |
48 |
730 |
483 |
110 |
4.95 |
730 |
6.09 |
573 |
304 |
71 |
5062,+ |
4.89 |
313 |
6.05 |
376 |
565 |
99 |
201 |
4.79 |
647 |
6.05 |
652* |
395* |
668 |
507+ |
4.76 |
510 |
6.01 |
395* |
760 |
781 |
365 |
4.64 |
652* |
6.01 |
780 |
318 |
57 |
653 |
4.55 |
15 |
6.01 |
455 |
7643 |
579 |
5022 |
4.52 |
814 |
6.01 |
5062,+ |
63 |
124 |
492 |
4.44 |
676 |
5.94 |
324 |
507+ |
590 |
456 |
4.43 |
782 |
5.94 |
Using PREDTAP
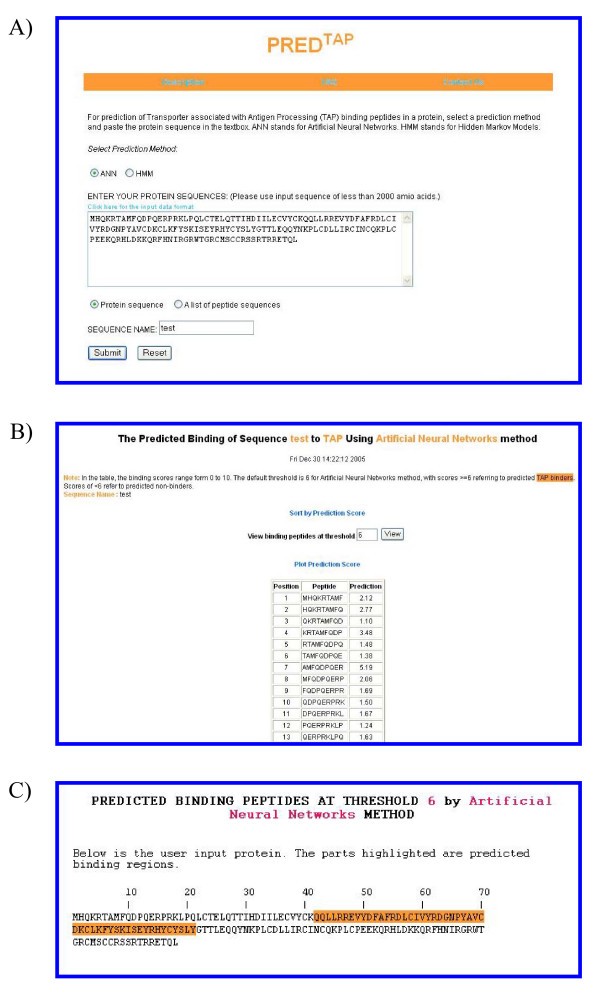
The examples of the output pages of PREDTAP for a single protein. The sequence type chosen is "protein sequence". A) The input page. B) The main result page. The input sequence is decomposed into overlapping 9-mers for prediction of binding scores to TAP. C) Alignment view of the predicted TAP binding regions in the input protein.
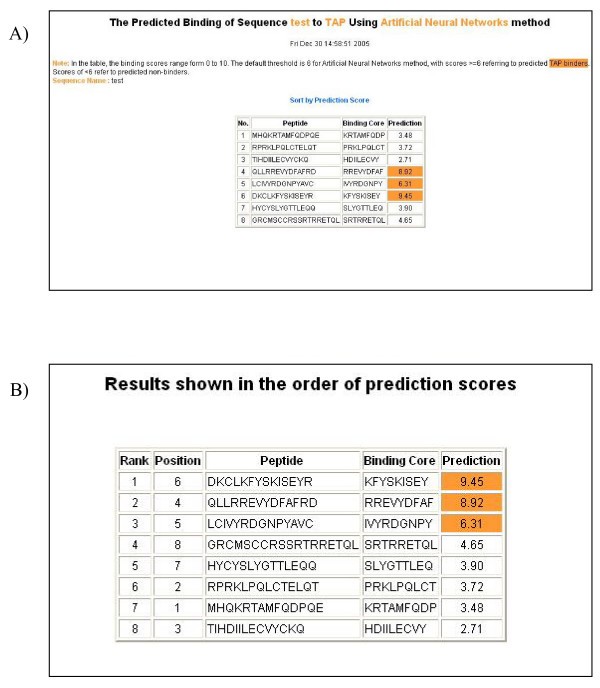
An example of the output pages of PREDTAP for a list of peptides. A) The input page. B) The main result page. All 9-mers in each peptide were submitted for prediction. The predicted binding scores are represented by the highest individual 9-mer binding score of each input peptide. The 9-mer with the highest binding score in each peptide is displayed as "Binding Core" in the result table.
Discussion
We have earlier compared four prediction servers for prediction of H-2Kd binding peptides [34]. A 121-amino acid long sequence of the nuclear export protein NS2 from influenza A virus (GenPept accession NP_859033) was searched for 9-mer candidate binders to a mouse MHC molecule H-2Kd using four internet-accessible systems. Only three peptides were predicted within the top ten candidates as binders by all four methods. The performance comparison of PREDTAP with SVMTAP and TAPPred (SVM) shows that consensus peptides can be selected by combining predictions. The examples suggested that individual predictions need to be taken with care and predictions may be improved by a consensus of multiple methods. A similar situation may be applicable to TAP predictions. Hence the combination of ANN and HMM predictions in PREDTAP should result in higher specificity (fewer false positives) at the cost of slightly lower sensitivity. The predictions by TAPPred (cascade SVM) appear to be of a limited value.
The combinatorial properties of molecular mechanisms involved in antigen processing and adaptive learning nature of the immune responses limit our ability to fully predict immune responses. Combining experimental and computational techniques improves our ability to decipher complex interactions of the immune system. Computer models are used to complement laboratory experiments and thereby speed up knowledge discovery in immunology. In particular, the number of large-scale laboratory experiments for T-cell epitope mapping can be minimised by the judicious use of experiments aimed at developing and validating computer models. These models can then be used to perform large-scale computer simulations rapidly and inexpensively. The hypotheses generated from these experiments can then be retested in the laboratory to confirm their applicability to real-life immunology. Further work will include both the refinement of computational models and scanning disease-related antigens for peptide sequences that show high probability of processing and presentation. Those peptides that are most likely to be produced by proteasomal cleavage, transported by TAP, and bound by HLA class I molecules are likely to be promising candidates for peptide-based CTL vaccines. The PREDTAP server provides for the prediction of peptide binding by TAP and can be used as a comparison method against other TAP-prediction servers.
Declarations
Acknowledgements
This project has been funded in part (GLZ, JTA, and VB) with the USA federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Grant No. 5 U19 AI56541 and U01 AI061142-01 and Contract No. HHSN266200400085C.
Authors’ Affiliations
References
- PRED TAP server [http://antigen.i2r.a-star.edu.sg/predTAP]
- Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA: Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev 2005, 207:145–157.View ArticlePubMedGoogle Scholar
- Strehl B, Seifert U, Kruger E, Heink S, Kuckelkorn U, Kloetzel PM: Interferon-gamma, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol Rev 2005, 207:19–30.View ArticlePubMedGoogle Scholar
- Saveanu L, Carroll O, Hassainya Y, van Endert P: Complexity, contradictions, and conundrums: studying post-proteasomal proteolysis in HLA class I antigen presentation. Immunol Rev 2005, 207:42–59.View ArticlePubMedGoogle Scholar
- Abele R, Tampe R: Function of the transport complex TAP in cellular immune recognition. Biochim Biophys Acta 1999, 1461:405–419.View ArticlePubMedGoogle Scholar
- Abele R, Tampe R: Modulation of the antigen transport machinery TAP by friends and enemies. FEBS Lett 2006, 580:1156–1163.View ArticlePubMedGoogle Scholar
- van Endert PM, Riganelli D, Greco G, Fleischhauer K, Sidney J, Sette A, Bach JF: The peptide-binding motif for the human transporter associated with antigen processing. J Exp Med 1995, 182:1883–1895.View ArticlePubMedGoogle Scholar
- Nijenhuis M, Schmitt S, Armandola EA, Obst R, Brunne J, Hammerling GJ: Identification of a contact region for peptide on the TAP1 chain of the transporter associated with antigen processing. J Immunol 1996, 156:2186–2195.PubMedGoogle Scholar
- Gadola SD, Moins-Teisserenc HT, Trowsdale J, Gross WL, Cerundolo V: TAP deficiency syndrome. Clin Exp Immunol 2000, 121:173–178.View ArticlePubMedGoogle Scholar
- Smith KD, Lutz CT: Peptide-dependent expression of HLA-B7 on antigen processing-deficient T2 cells. J Immunol 1996, 156:3755–3764.PubMedGoogle Scholar
- Brusic V, Bajic VB, Petrovsky N: Computational methods for prediction of T-cell epitopes – a framework for modelling, testing, and applications. Methods 2004, 34:436–43.View ArticlePubMedGoogle Scholar
- Daniel S, Brusic V, Caillat-Zucman S, Petrovsky N, Harrison L, Riganelli D, Sinigaglia F, Gallazzi F, Hammer J, van Endert PM: Relationship between peptide selectivities of human transporters associated with antigen processing and HLA class I molecules. J Immunol 1998, 161:617–624.PubMedGoogle Scholar
- Peters B, Bulik S, Tampe R, Van Endert PM, Holzhutter HG: Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol 2003, 171:1741–1749.PubMedGoogle Scholar
- Doytchinova I, Hemsley S, Flower DR: Transporter associated with antigen processing preselection of peptides binding to the MHC: a bioinformatic evaluation. J Immunol 2004, 173:6813–6819.PubMedGoogle Scholar
- Brusic V, van Endert P, Zeleznikow J, Daniel S, Hammer J, Petrovsky N: A neural network model approach to the study of human TAP transporter. In Silico Biol 1999, 1:109–121.PubMedGoogle Scholar
- Bhasin M, Raghava GP: Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci 2004, 13:596–607.View ArticlePubMedGoogle Scholar
- Petrovsky N, Brusic V: Virtual models of the HLA class I antigen processing pathway. Methods 2004, 34:429–35.View ArticlePubMedGoogle Scholar
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M: An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol 2005, 35:2295–2303.View ArticlePubMedGoogle Scholar
- Donnes P, Kohlbacher O: Integrated modeling of the major events in the MHC class I antigen processing pathway. Protein Sci 2005, 14:2132–2140.View ArticlePubMedGoogle Scholar
- Peters B, Sette A: Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 2005, 6:132.View ArticlePubMedGoogle Scholar
- Tenzer S, Peters B, Bulik S, Schoor O, Lemmel C, Schatz MM, Kloetzel PM, Rammensee HG, Schild H, Holzhutter HG: Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci 2005, 62:1025–1037.View ArticlePubMedGoogle Scholar
- Doytchinova IA, Guan P, Flower DR: EpiJen: a server for multistep T cell epitope prediction. BMC Bioinformatics 2006, 7:131.View ArticlePubMedGoogle Scholar
- Groothuis TA, Griekspoor AC, Neijssen JJ, Herberts CA, Neefjes JJ: MHC class I alleles and their exploration of the antigen-processing machinery. Immunol Rev 2005, 207:60–76.View ArticlePubMedGoogle Scholar
- Lautscham G, Mayrhofer S, Taylor G, Haigh T, Leese A, Rickinson A, Blake N: Processing of a multiple membrane spanning Epstein-Barr virus protein for CD8(+) T cell recognition reveals a proteasome-dependent, transporter associated with antigen processing-independent pathway. J Exp Med 2001, 194:1053–1068.View ArticlePubMedGoogle Scholar
- Zimmer J, Andres E, Donato L, Hanau D, Hentges F, de la Salle H: Clinical and immunological aspects of HLA class I deficiency. QJM 2005, 98:719–727.View ArticlePubMedGoogle Scholar
- Brusic V, Petrovsky N, Zhang G, Bajic VB: Prediction of promiscuous peptides that bind HLA class I molecules. Immunol Cell Biol 2002, 80:280–285.View ArticlePubMedGoogle Scholar
- Udaka K, Mamitsuka H, Nakaseko Y, Abe N: Empirical evaluation of a dynamic experiment design method for prediction of MHC class I-binding peptides. J Immunol 2002, 169:5744–5753.PubMedGoogle Scholar
- Zhang P: Model selection via multifold cross validation. Ann Stat 1993, 21:299–313.View ArticleGoogle Scholar
- Swets JA: Measuring the accuracy of diagnostic systems. Science 1988, 240:1285–1293.View ArticlePubMedGoogle Scholar
- Kast WM, Brandt RM, Sidney J, Drijfhout JW, Kubo RT, Grey HM, Melief CJ, Sette A: Role of HLA-A motifs in identification of potential CTL epitopes in human papillomavirus type 16 E6 and E7 proteins. J Immunol 1994, 152:3904–3912.PubMedGoogle Scholar
- Monji M, Nakatsura T, Senju S, Yoshitake Y, Sawatsubashi M, Shinohara M, Kageshita T, Ono T, Inokuchi A, Nishimura Y: Identification of a novel human cancer/testis antigen, KM-HN-1, recognized by cellular and humoral immune responses. Clin Cancer Res 2004, 10:6047–6057.View ArticlePubMedGoogle Scholar
- Smith KD, Lutz CT: Peptide-dependent expression of HLA-B7 on antigen processing-deficient T2 cells. J Immunol 1996, 156:3755–3764.PubMedGoogle Scholar
- Neisig A, Roelse J, Sijts A, Ossendorp F, Feltkamp M, Kast W, Melief C, Neefjes J: Major differences in transporter associated with antigen presentation (TAP)-dependent translocation of MHC class I-presentable peptides and the effect of flanking sequences. J Immunol 1995, 154:1273–1279.PubMedGoogle Scholar
- Brusic V, Petrovsky N: Immunoinformatics and its relevance to understanding human immune disease. Expert Rev Clin Immunol 2005, 1:145–157.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.