
In silico identification of opossum cytokine genes suggests the complexity of the marsupial immune system rivals that of eutherian mammals
- Research
- Open Access
In silico identification of opossum cytokine genes suggests the complexity of the marsupial immune system rivals that of eutherian mammals
- Emily SW Wong1_10Email author,
- Lauren J Young2_10Email author,
- Anthony T Papenfuss3_10 and
- Katherine Belov1_10Email author
- Received: 26 June 2006
- Accepted: 10 November 2006
- Published: 10 November 2006
Abstract
Background
Cytokines are small proteins that regulate immunity in vertebrate species. Marsupial and eutherian mammals last shared a common ancestor more than 180 million years ago, so it is not surprising that attempts to isolate many key marsupial cytokines using traditional laboratory techniques have been unsuccessful. This paucity of molecular data has led some authors to suggest that the marsupial immune system is 'primitive' and not on par with the sophisticated immune system of eutherian (placental) mammals.
Results
The sequencing of the first marsupial genome has allowed us to identify highly divergent immune genes. We used gene prediction methods that incorporate the identification of gene location using BLAST, SYNTENY + BLAST and HMMER to identify 23 key marsupial immune genes, including IFN-γ, IL-2, IL-4, IL-6, IL-12 and IL-13, in the genome of the grey short-tailed opossum (Monodelphis domestica). Many of these genes were not predicted in the publicly available automated annotations.
Conclusion
The power of this approach was demonstrated by the identification of orthologous cytokines between marsupials and eutherians that share only 30% identity at the amino acid level. Furthermore, the presence of key immunological genes suggests that marsupials do indeed possess a sophisticated immune system, whose function may parallel that of eutherian mammals.
Keywords
- Eutherian Mammal
- Monodelphis Domestica
- Chicken Sequence
- Devil Facial Tumour Disease
- Opossum Genome
Background
The marsupial and eutherian (placental) lineages diverged approximately 180 million years ago. Marsupials are chiefly distinguished from other mammals by their unique reproductive strategies, with young born in an immature state with only the most rudimentary neurological and immunological systems [1]. At birth, the animal manoeuvres its way to a waiting teat, where it attaches until it reaches a state of maturity that allows it to function independently. Marsupials possess lymphoid tissue and cellular components that are structurally similar to those of other mammals. Key antigen receptor and recognition molecules including Major Histocompatibility (MHC) Class I, II and III [2], T Cell Receptors alpha, beta, gamma and delta [3, 4], Toll-like receptors [5] and immunoglobulins [6] have been characterized.
However, conventional experimental strategies using degenerate primers for reverse-transcriptase polymerase chain reaction (RT-PCR) and heterologous probes for screening genetic libraries have only identified the most phylogenetically conserved immune molecules, with cytokines proving particularly difficult to isolate [7]. To date, only eleven cytokines including one receptor have been cloned from marsupials. They include tumour necrosis factor alpha (TNF-α) [8, 9], lymphotoxin (LT) -α and -β [10, 11], Interleukin IL-1β [12], IL-1R2 [7], IL-5 [13], IL-10 [14], leukemia inhibitory factor LIF; a member of the IL-6 family [15] and three type I Interferon (IFN) genes [16]. These cytokines show relatively high levels of identity compared to their eutherian homologues. Previous attempts to isolate the more divergent T-cell derived cytokines that orchestrate adaptive immunity such as IL-2, IL-4 and interferon-γ have failed [7, 17].
Identification of divergent marsupial immune genes is important for two reasons. Firstly, unsuccessful attempts to isolate T cell derived cytokines in the laboratory has led some authors to suggest that the marsupial immune system is 'primitive' and does not possess the level of complexity demonstrated by eutherians such as humans and mice. The fact that some T cell driven responses are also aberrant adds to this argument. Marsupials appear to have delayed skin graft rejection [18] and antibody class switching [19], together with an apparent lack of an in vitro Mixed Lymphocyte Response [20]. Elucidation of genes involved in specific immunity will help us to determine whether the apparently 'simple' immune responses generated by marsupials are genetically hardwired.
The second reason for identifying divergent immune genes in the marsupial genome is to develop marsupial specific immunological reagents. To date, most assay systems employed to characterise cells and their function rely on eutherian reagents or culture techniques developed in eutherian species. Where low levels of cross reactivity exist between marsupials and these model species, the usefulness of the data generated from such assays is limited. Identification of key cell markers, such as CD4 and CD8 will allow us to generate marsupial-specific reagents, which would then be used to gain a better understanding of the marsupial immune response.
Difficulties associated with identifying rapidly evolving cytokines are not limited to marsupials. The chicken IL-2 gene took seven years of focused effort to identify [21], and was eventually found using expression strategies and not heterologous cloning techniques. The recent sequencing of the complete genomes of a large number of non-eutherian vertebrates will expedite the isolation and characterization of these immune genes in distantly related species. However, currently automated annotation techniques are not sensitive enough to identify many of these molecules outside the eutherian lineage.
The first marsupial genome was recently sequenced by the Broad Institute. The subject of this project, Monodelphis domestica, is a South American opossum. It is a well-recognised biomedical model in the study of comparative physiology, immunogenetics, cancer development and disease susceptibility. Two publicly available annotations of this genome have been generated. Ensembl have produced a gene build with their automatic pipeline [22], which relies principally on GeneWise [23], while the UCSC genome browser provides several annotation tracks with similarity features and gene models, for example chained TBLASTN alignments of human proteins, BLAT alignments of RefSeq mRNAs, and Genscan [24] and N-SCAN [25] predictions. With the exception of the Genscan predictions, which are ab initio gene predictions based on genomic sequence only, the gene builds rely on cross species homology, as no large-scale opossum EST projects have been completed yet and there are only 425 known opossum protein sequences in GenBank. In most cases, Ensembl and the UCSC genome browser were unable to identify highly divergent cytokine genes such as IL-2, 4 and 13.
To overcome this shortcoming in the automated annotation of the opossum genome and to start to address uncertainties about immune function in marsupials, we have adopted a manual, expert-curated approach to annotating highly divergent genes. The critical first stage of this is the careful identification of the genomic region containing the gene. This is performed using a sensitive TBLASTN search. HMMER [26] can also be useful at this stage. Frequently, it is necessary to first narrow the search to the syntenic region by identifying conserved flanking genes.
Having identified similarity features, gene prediction is performed on genomic sequence extracted from the region. The accuracy of gene prediction is dependent on the prediction method. As with the automated annotations, we favour gene predictors that incorporate information from orthologous sequences into the prediction process. In addition to GeneWise and N-Scan, there are now several such methods available including Procrustes [27], HMMgene,[28] GenomeScan [29], and Augustus+ [30]. Procrustes and the default GeneWise algorithm perform spliced alignment. Augustus+ uses an interesting approach, which constrains predicted genes to incorporate user-supplied hints. However, it is not particularly convenient for manual use or use by biologists lacking scripting skills. While not the only possible choice, we have found GenomeScan to be both convenient and reasonably accurate (based on comparison with known eutherian sequences). It is worth noting that there is another class of gene prediction methods that obtain homology information from syntenic regions of other genomes. These include TwinScan [31], which is asymmetric and predicts genes in one genome only and SLAM [32], which simultaneously aligns two genomes and predicts genes in both. These methods were unlikely to be useful in our study as we were looking for genes that are highly divergent and difficult to align at the genomic level. Finally, a comparison of predicted results with known eutherian sequences and curation of the result was undertaken if required. Our success with this strategy suggests that this method will be applicable to the identification of rapidly evolving gene families in other distant vertebrate species.
Results
Overview
Comparison of putative opossum and known human cytokine sequences. Opossum IFN-α genes were compared with 13 human IFN-α genes.
Identity |
Number of exons in open reading frame |
Number of amino acids |
% amino acid identity* |
||
|---|---|---|---|---|---|
Cytokines |
opossum |
human |
opossum |
human |
|
Interleukin 2 |
4 |
4 |
144 |
153 |
41.8 |
Interleukin 4 |
4 |
4 |
138 |
153 |
43.3 |
Interleukin 5 |
4 |
4 |
137 |
134 |
53.0 |
Interleukin 6 |
5 |
5 |
221 |
212 |
36.3 |
Interleukin 10 |
5 |
5 |
173 |
178 |
59.5 |
Interleukin 12A |
7 |
7 |
231 |
253 |
52.2 |
Interleukin 13 |
4 |
4 |
122 |
146 |
36.7 |
Interleukin 19 |
6 |
6 |
188 |
215 |
58.0 |
Interleukin 20 |
6 |
5 |
154 |
176 |
59.7 |
Interleukin 21 |
5 |
5 |
163 |
162 |
46.5 |
Interleukin 22 |
4 |
5 |
150 |
179 |
40.0 |
Interleukin 24 |
6 |
6 |
220 |
206 |
43.7 |
Interleukin 26 |
5 |
5 |
201 |
171 |
55.9 |
Interferon γ |
4 |
4 |
167 |
166 |
47.0 |
Type 1 Interferons |
|||||
IFN-α (seven genes) |
1 |
1 |
183 |
188–9 (range) |
33–42 (range) |
IFN-β |
1 |
1 |
184 |
187 |
43.2 |
IFN-κ |
1 |
1 |
156 |
207 |
51.3 |
Cytokine receptors |
|||||
Common cytokine receptor gamma chain (IL-2Rγ) |
8 |
8 |
349 |
367 |
54.9 |
Interferon-γ receptor 2 |
6 |
7 |
269 |
344 |
49.6 |
T cell surface receptors |
|||||
CD4 |
9 |
9 |
485 |
458 |
45.6 |
CD8 |
6 |
6 |
349 |
235 |
37.7 |
Summary of putative opossum cytokine genes including search strategy, best hit, predicted glycosylation sites and signal peptide information.
Identity |
Search Strategy |
Best Hit |
Number of predicted glycosylation sites |
Signal peptide identified |
||
|---|---|---|---|---|---|---|
Cytokines |
reference |
e-value |
N-gly |
O-gly |
opossum |
|
Interleukin 2 |
synteny |
- |
- |
1 |
1 |
Yes |
Interleukin 4 |
synteny |
- |
- |
2 |
0 |
Yes |
Interleukin 5 |
BLAST |
AAD37462.1 |
4e-028 |
1 |
0 |
Yes |
Interleukin 6 |
BLAST |
NP_112445.1 |
0.081 |
2 |
3 |
Yes |
Interleukin 10 |
BLAST |
AAD01799.1 |
3e-011 |
2 |
0 |
Yes |
Interleukin 12A |
BLAST |
NP_032377.1 |
9e-006 |
1 |
1 |
No |
Interleukin 13 |
synteny/HMMER |
- |
- |
0 |
0 |
No |
Interleukin 19 |
BLAST |
NP_001009940.1 |
3e-008 |
3 |
0 |
Yes |
Interleukin 20 |
BLAST |
NP_061194.2 |
1e-004 |
0 |
0 |
Yes |
Interleukin 21 |
BLAST |
Q9HBE4 |
0.088 |
2 |
1 |
Yes |
Interleukin 22 |
BLAST |
NP_065386.1 |
3e-005 |
1 |
0 |
Yes |
Interleukin 24 |
BLAST |
NP_006841.1 |
2e-006 |
2 |
0 |
Yes |
Interleukin 26 |
BLAST |
NP_060872.1 |
3e-009 |
0 |
0 |
No |
Interferon-γ |
synteny |
- |
- |
2 |
0 |
Yes |
Type 1 Interferons |
||||||
IFN-α (7 genes) |
BLAST |
AAO37656.1 |
1e-019 to 9e-974 (range) |
0–4 (range) |
0 |
Yes |
IFN-β |
BLAST |
AAO37656.1 |
2 |
0 |
Yes |
|
IFN-κ |
BLAST |
AAO37656.1 |
1 |
0 |
No |
|
Cytokine receptors |
||||||
Common cytokine receptor gamma chain (IL-2Rγ) |
BLAST |
NP_000197.1 |
9e-019 |
6 |
3 |
No |
Interferon-γ receptor 2 |
BLAST |
NP_005525.2 |
3e-018 |
3 |
2 |
Yes |
T cell surface receptors |
||||||
CD4 |
BLAST |
NP_000607.1 |
5e-009 |
4 |
0 |
Yes |
CD8 |
BLAST |
Q60965 |
9e-012 |
1 |
8 |
Yes |
Isolation of interleukins using BLAST and synteny searches
Interleukins 2, 4 and 21 and their common gamma chain receptor were identified using both BLAST and syntenic strategies. IL-21 was identified by a sensitive TBLASTN search (e-value = 2e-18) on Chromosome 5:7034081–7057815. The predicted protein is of similar size and contains the same number of exons as human IL-21 [see Additional File 1]. The signal peptide was predicted to be encoded within the first 21 amino acids (score = 7.6, p = 0.06), with N-linked glycosylation sites predicted at positions 46 and 106 and O-linked glycosylation of threonine predicted at position 55. Instability motifs (ATTTA) were not found in the 3' UTR of the sequence before the poly(A)+ signal.
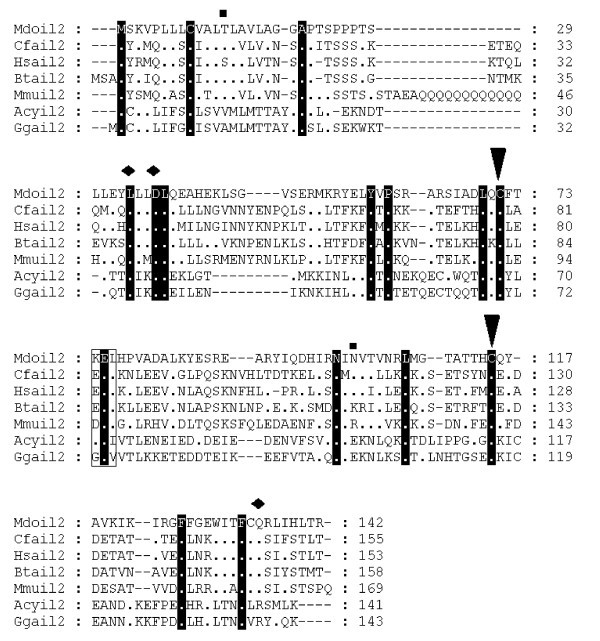
Alignment of IL-2 amino acid sequences. Diamonds denote functionally important residues [64]. Inverted triangles indicate cysteine residues involved in disulfide bonds in human protein [64]. Squares above the alignment show predicted glycosylation sites from the opossum sequence. Dots represent identity to Monodelphis domestica sequence. Sequences used for alignment: Homo sapiens (NP_000577), Bos taurus (NP_851340), Sus scrofa (NP_999026), Mus musculus (NP_032392), Gallus gallus (NP_989484), Canis familiaris (NP_001003305), Macaca fascicularis (Q29615), Felis cattus (AAC15974), Equus caballus (CAA49190), Cervus elaphus (P51747), Capra hircus (AAQ10671), Ovis aries (NP_001009806), Oryctolagus cuniculus (O77620), Peromyscus maniculatus (AAP04419), Rattus norvegicus (NP_446288), Anser cygnoides (AAR28994). Not all sequences are shown in the figure.
Opossum IL-2Rγ was identified using TBLASTN (e-value = 8e-119) (Table 1). It shares 61% amino acid similarity with the human sequence [see Additional File 3].
IL-5 was identified on chromosome 1:307529660–307531352. It shares 53.0% identity to human IL-5, and 86.7% identity to the tammar wallaby IL-5 [13] [see Additional File 4].

Alignment of IL-4 amino acid sequences. Inverted triangles indicate cysteine residues that form disulfide bonds in the human protein [65]. Squares above the alignment show predicted N-linked glycosylation sites in the opossum sequence. Dots represent identity to Monodelphis domestica sequence. Sequences used for alignment: Homo sapiens (NP_000580), Mus musculus (NP_067258), Gallus gallus (NP_001007080), Equus caballus (P42202), Rattus norvegicus (NP_958427), Mesocricetus auratus (Q60440).
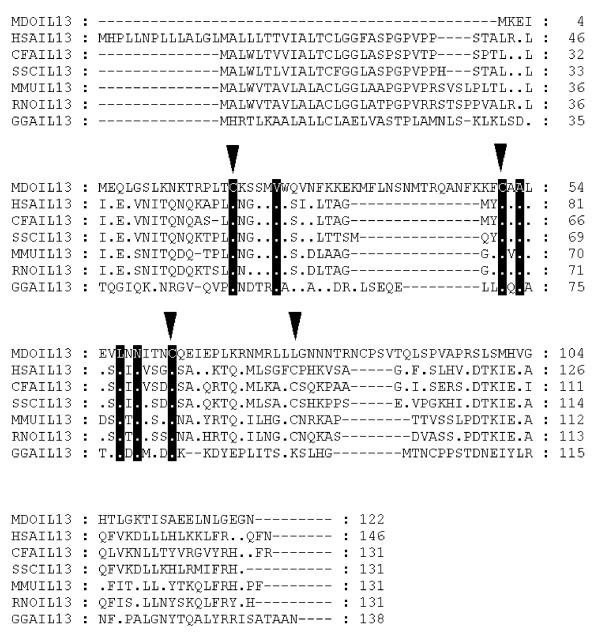
Alignment of IL-13 amino acid sequences. Inverted triangles indicate cysteine residues that form disulfide bonds in the human protein [66]. Dots represent identity to Monodelphis domestica sequence. Sequences used for alignment: Homo sapiens (NP_002179), Mus musculus (NP_032381), Gallus gallus (NP_001007086), Rattus norvegicus (NP_446280), Canis familiaris (NP_001003384), Sus scrofa (Q95J68).
Opossum IL-6 was identified using a sensitive TBLASTN search (e-value = 0.08). Opossum IL-6 is located on Chromosome 8:296810942–296824133 and the PROSITE IL-6 family motif (C-x(9)-C-x(6)-G-L-x(2)- [F,Y]-x(3)-L) is conserved [see Additional File 6]. The signal peptide is predicted from positions 1–28 (score = 8.1, p = 0.20) and no instability motifs (ATTTA) are found in the 3' UTR. Opossum IL-6 has maintained significant structural similarities to human and other mammalian IL-6 genes despite its comparatively low sequence identity. The number and position of cysteine residues in opossum IL-6 are identical to those found in eutherian and chicken sequences. An arginine molecule in helix D that is involved in IL-6β binding [38] is also conserved.
Opossum IL-12 alpha chain (chr7:260,616,009–260,626,803) was identified using a TBLASTN search and is predicted to be 58% similar to its human orthologue [see Additional File 7]. Cysteine residues are conserved between the marsupial, eutherians and chicken sequences.
IL-10 family members were identified in two clusters. Chromosome 2 contained IL-10 (113139397–113144942; [see Additional File 8]), IL-19 (113283404–113294773; [see Additional File 9]), IL-20 (113319666–113324608; [see Additional File 10]), IL-24 (113362216–113377467; [see Additional File 11]) with identical head-to-tail transcriptional orientation and organisation to their human orthologues. Chromosome 8 contained IL-26 (23485674–23494985; [see Additional File 12]) and IL-22 (23457582–23460076; [see Additional File 13]). The complete IL-22 open reading frame was not identified since the 3' end (approximately 33 amino acids and 2 exons) fell in an unsequenced gap. However, conservation of a predicted N-linked glycosylation site at N54 between putative opossum IL-22 and human IL-22 (a site crucial for IL-22 modulation during the inflammatory response) suggests that this partial sequence is opossum IL-22. Both chicken and the amphibia contain IL-10 family members, although only one IL-19-like ancestral gene replaces IL-19, IL-20 and IL-24 in the chicken [39]. Orthology of the IL-10 family cytokines with their eutherian counterparts was confirmed by phylogenetic analysis [see Additional File 14]. All putative IL-10 family members clustered closely with their eutherian orthologs.
Isolation of cluster of differentiation markers using TBLASTN
CD4 [see Additional File 15] and CD8 [see Additional File 16] were identified by TBLASTN search and found on chromosome 8 (104157682–104183462) and chromosome 1 (716671734–716675645) respectively. Their number of amino acids and potential glycosylation sites are noted in Table 2. Neither we, nor Ensembl, were able to successfully predict the terminal exons of these two genes.
Isolation of interferons using BLAST, synteny and hidden Markov models
Type I IFNs
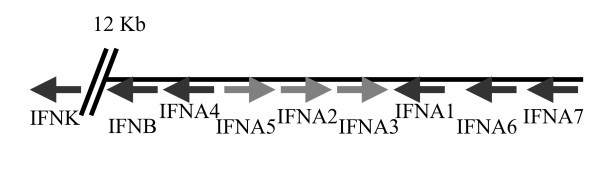
Genomic organisation and transcriptional directions of type I IFNs on chromosome 6.
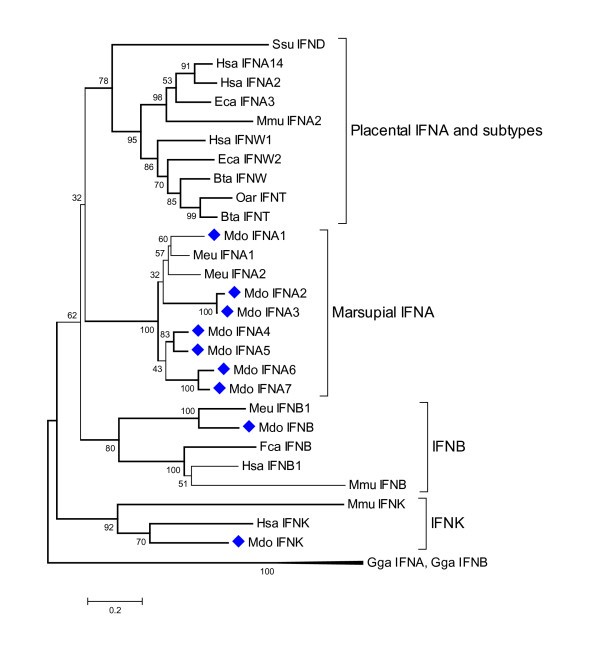
Phylogenetic tree showing evolutionary relationship between type I interferon protein sequences. Opossum sequences are marked by diamonds. Sequences used: Homo sapiens IFN-α14 (NP_002163.1), IFN-α2 (NP_000596.2), IFN-β (NP_002167), IFN-ω1 (P07352), IFN-κ(NP_064509.1); Mus musculus IFN-α2 (P01573), IFN-β (NP_034640), IFN-κ (NP_954608.1); Sus scrofa IFN-δ (NP_001002832.1); Equus caballus IFN-α3 (CAA01292), IFN-ω2 (CAA01293); Bos taurus IFN-ω1 (P07352), IFN-t (XP_874910); Ovis aries IFN-τ(CAA39783); Felis catus IFN-β (Q9N2J0); Macropus eugenii IFN-α1 (AAO37656), IFN-α2 (AAO37657.1), IFN-β (AAO37658.1); Gallus gallus IFN-α1 (CAA63214), IFN-β (NP_001020007).
Interferon gamma (IFN-γ) and interferon gamma receptor (IFN-γR2)

Alignment of IFN-γ amino acid sequences. Squares above the alignment show predicted N-linked glycosylation sites from the opossum sequence. Dots represent identity to Monodelphis domestica sequence. Sequences used for alignment: Homo sapiens (NP_000610.2), Felis cattus (P46402), Equus caballus (P42160), Equus asinus (O77763), Sus scofa (NP_999113.1), Ovis aries (P17773), Mus musculus (NP_032363.1), Rattus norvegicus (NP_620235.1), Peromyscus maniculatus (AAP44086.1), Gallus gallus (P49708), Danio rerio (NP_998029.1).
Level of confidence in our gene predictions
Where possible, gene predictions were verified by alignment with known marsupial cDNA sequences, and compared to Ensembl gene predictions and UCSC similarity features. For instance, a known cDNA sequence is available for Trichosurus vulpecula (possum) IL-10 cDNA (Genbank ref: AF026277). Our predicted opossum IL-10 protein shared 76% amino acid identity with possum IL-10, and exon-intron boundaries match. However, despite our use of robust methodologies, we are still not confident with prediction of the most divergent immune genes sequences. Some doubt exists with our predications for IL-4 and IL-13 and the terminal exons of IL-22, CD4 and CD8. Characterisation of their cDNA, together with laboratory-based assays will ultimately confirm the reliability of the predictions reported here.
Discussion
Without EST and protein databases, annotation of distantly related mammalian species such as the marsupials and monotremes is challenging. Neither Ensembl nor UCSC were able to identify IL-2, 4, 13, 22 and IFN-γ. In general, automated gene prediction missed key immune genes because of their low levels of sequence similarity with their eutherian orthologs. We suggest that future studies focusing on in silico mining of divergent genes should take into account gene location and features. Application of this strategy allowed us to successfully identify key immune genes in the opossum genome, which traditional laboratory methods failed to isolate.
Discovery of key cytokines in the opossum genome suggests that a re-examination of immune responses (especially T cell responses) is warranted in marsupials. The peculiarities in class switching and in vitro T cell proliferation, which have previously been observed in marsupials are largely controlled by T cells and their products. The ability to discriminate between classic 'helper' T cells and 'cytotoxic' T cell families will now be possible due to the identification of CD4 and CD8 sequences in the opossum genome. Further, identification of cytokines normally produced by these subsets in eutherian mammals will allow us to investigate Th1 and Th2 profiles that orchestrate immunity to intracellular and extracellular pathogens respectively.
There are a myriad of interactions between cytokines at the cellular level, but the presence of a number of key cytokines orchestrate the global immune response. For example, when the macrophage-derived IL-12 is dominant, Th1 responses predominate resulting in cell-mediated immunity. When the B-cell growth factor IL-4 is dominant, Th2 responses dominate and a humoral immune response is activated [40]. Sequences for both of these genes are present in the opossum genome, along with other classical Th1- (IL-2, IFN-γ) and Th2- (IL-4, IL-5, IL-6, IL-10 and IL-13) associated molecules.
The presence of key cytokines in a marsupial genome strongly suggests that marsupials are capable of complex immune responses comparable to those seen in eutherian mammals. Knowledge of these gene sequences provides a springboard for future studies. For instance, marsupials appear to be susceptible to infection with intracellular pathogens such as herpesvirus and mycobacterial spp [41], indicative of impaired Th1 cytokine responses. The availability of Th1 and Th2 cytokine sequences will allow us to study IL-10 profiles, which are known to play a critical role in the survival of intracellular pathogens by inhibiting the expression of inflammatory cytokines such as IFN-γ and TNF. Meanwhile, studies of Th2 cytokines may focus on protection against parasites. Both American and Australian marsupials co-exist with a range of successful parasites; opossums are reported to have natural trypanosome infection rates of up to 100% [42] and carry nematode burdens in the wild [43], whilst a variety of helminth infections are common across a range of Australian marsupials [44].
Allograft responses can now be studied due to the availability of sequence information for interleukins 2, 4, 21 and IL-2Rγ [45]. The opossum is an important model for tumour immunology since it can be induced to accept melanoma cells at both juvenile and adult life stages [46]. Both IL-2 and IL-24 are associated with melanoma tumour suppression [47] in humans and it is now possible to study the role of these genes in the opossum model, as well as in the maintenance of transmissible allograft tumours in Tasmanian devil facial tumour disease [48].
Conclusion
Here we describe and apply a method to identify divergent immune genes from the genome of a model marsupial, Monodelphis domestica. We are now extending this analysis to characterize the entire opossum immunome. We report here that the opossum genome contains representatives from the major vertebrate immune gene families. These genes appear to be structurally similar, and therefore will most likely prove to be functionally equivalent, to their eutherian homologues. The way is now clear to further probe the genes that orchestrate the marsupial immune response and to investigate the role that these molecules have on maintaining health and influencing disease susceptibility in this unique group of animals.
Methods
Data source
Draft sequencing of the genome of a female opossum (Monodelphis domestica) has recently been completed by the Broad Institute [49]. Analysis was performed on assembly MonDom4 (January 2006).
Sequence identification
To optimise the chances of identifying previously undiscovered sequences, our search strategy relied on a preliminary database screen for sequence conservation, together with positional analyses of the gene sequence relative to other genes within the genome (synteny). Finally, the putative sequence was analysed for the presence of biologically significant sites associated with both structure and function in their eutherian homologues.
Similarity searching using BLAST
Sequence similarity searching (TBLASTN) was performed with known eutherian sequences. Positive hits from the BLAST search with good potential were extracted for further structural analysis. When ambiguities existed between alignments from BLAST results, each of the multiple hits were extracted and inspected. Assessment methods for ambiguous hits included tests for reciprocal-best-hit where the aligned sequence was blasted against SWISS-PROT and TrEMBL protein databases to confirm preliminary findings. Proteins discovered in BLAST searches were used to mine additional homologues. To do this, parameters were optimised for sensitive searching. In order to increase our ability to detect highly divergent sequences, the BLOSUM 45 similarity matrix [50] was used. Additionally, application of soft-masking and the lowering of the neighbourhood word threshold score to 9 increased the chance of detecting homologous sequences that otherwise might have been overlooked using default parameters.
Synteny analysis
If the protein of interest was not detected by the initial BLAST search, other methods were employed. Similarity searches were performed with genes found in close syntenic regions in the human genome. Syntenic regions were extracted from the opossum database, and passed into GENSCAN [24]. The predicted peptide sequences were analysed by performing similarity searches against the SWISS-PROT and TrEMBL databases using BLASTP and FASTP [51]. In order to improve the accuracy of identified cytokine sequences, the sequence was re-extracted from the opossum database and the putative protein was re-evaluated using GenomeScan [29]. Combined results from GenomeScan and GENSCAN were compared with documented structural features of the cytokine.
Additional methods for gene identification
For sequences that were not detected using the above methods, a hidden Markov model (HMM) was built and calibrated using the HMMER 2.3.2 package [52]. The model was built as a multiple local alignment profile with the Krogh/Mitchison substitution weight matrix [53] and used to search the six-frame translation of the opossum genome.
Ancestral sequences were included in the HMM. These were calculated by programs in the Phylip package [54]. PRODIST was used to compute a distance matrix under default settings. After this, the program NEIGHBOUR was used to create a neighbour joining (NJ) tree from the matrix. The tree was rooted with a teleost species. Following this, ProML was set to produce ancestral sequences at each of the NJ tree nodes.
Structural features
Once the gene of interest was located, exon/intron boundaries were identified using the gene prediction programs GENSCAN [24] and GenomeScan [29]. Our experience suggests that some caution is advisable in the interpretation of data from existing gene prediction software; excessively long predicted genes ('thready' gene predictions) due to mis-identification of first exons and merging of adjacent genes, and unlikely predictions of splice sites (based on comparison with orthologous sequences) were the most common problems we observed. Mindful of these limitations, our gene predictions were compared with known gene structures. The presence of signal peptides was predicted by SPScan (Accelrys GCG) and estimation of glycosylation sites were made with NetOGlyc 3.1 [55] and NetNGlyc 1.0 [56]. Finally, sequences were submitted to the PROSITE database [57] for detection of protein family motifs that would confirm gene identify.
Sequence alignments
Sequences from the opossum and other species were aligned using ClustalW [58]. Accession numbers of sequences used in analyses are shown in figure legends. Sequence labels in the alignments are abbreviated by the first letter from the genus with the first two letters from the species name followed by the gene name. In figures, residues with functional importance are highlighted.
Phylogenetic analysis
Neighbour-joining (NJ) trees were constructed using the Jones-Taylor-Thornton substitution model [59] and 500 bootstrap replicates in MEGA 3.1 [60]. The tree, constructed from amino acid sequences, was rooted using chicken sequences.
Sequence identity
Sequence identity and similarity calculations were carried out using GAP (Accelrys GCG), with the Needleman-Wunsch alignment [61], except for IFN-α genes which were calculated in GenDoc [62] using the BLOSUM 35 similarity matrix [50] for comparisons of human and opossum IFN-α genes and BLOSUM 80 matrix [50] for comparisons among opossum sequences. GCG, GENSCAN, BLASTP and FastA programs were accessed through the Australian National Genomic Information Service (ANGIS) [63].
Declarations
Acknowledgements
This work was funded by the Australian Research Council (KB), Central Queensland University (LJY) and the University of Sydney (KB). EW's PhD scholarship is funded by the ARC Centre for Kangaroo Genomics and the Jean Walker Trust. We thank the Broad Institute (especially Kerstin Lindblad-Toh and April Cook) for providing us with early access to the opossum genome data and for sequencing an additional BAC which was important for this study. We also gratefully acknowledge the encouragement and support of Terry Speed, who contributed to the publication costs of this manuscript.
Authors’ Affiliations
References
- Tyndale-Biscoe H: Life of Marsupials. Victoria, Australia, CSIRO Publishing 2005.Google Scholar
- Belov K, Deakin JE, Papenfuss AT, Baker ML, Melman SD, Siddle HV, Gouin N, Goode DL, Sargeant TJ, Robinson MD, Wakefield MJ, Mahony S, Cross JG, Benos PV, Samollow PB, Speed TP, Graves JA, Miller RD: Reconstructing an ancestral mammalian immune supercomplex from a marsupial major histocompatibility complex. PLoS Biol 2006, 4:e46.View ArticlePubMedGoogle Scholar
- Baker ML, Osterman AK, Brumburgh S: Divergent T-cell receptor delta chains from marsupials. Immunogenetics 2005, 57:665–673.View ArticlePubMedGoogle Scholar
- Baker ML, Rosenberg GH, Zuccolotto P, Harrison GA, Deane EM, Miller RD: Further characterization of T cell receptor chains of marsupials. Dev Comp Immunol 2001, 25:495–507.View ArticlePubMedGoogle Scholar
- Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A: The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A 2005, 102:9577–9582.View ArticlePubMedGoogle Scholar
- Miller RD, Belov K: Immunoglobulin genetics of marsupials. Dev Comp Immunol 2000, 24:485–490.View ArticlePubMedGoogle Scholar
- Harrison GA, Wedlock DN: Marsupial cytokines. Structure, function and evolution. Dev Comp Immunol 2000, 24:473–484.View ArticlePubMedGoogle Scholar
- Harrison GA, Broughton MJ, Young LJ, Cooper DW, Deane EM: Conservation of 3' untranslated region elements in tammar wallaby (Macropus eugenii) TNF-alpha mRNA. Immunogenetics 1999, 49:464–467.View ArticlePubMedGoogle Scholar
- Wedlock DN, Aldwell FE, Buddle BM: Molecular cloning and characterization of tumor necrosis factor alpha (TNF-alpha) from the Australian common brushtail possum, Trichosurus vulpecula. Immunol Cell Biol 1996, 74:151–158.View ArticlePubMedGoogle Scholar
- Harrison GA, Deane EM: cDNA sequence of the lymphotoxin beta chain from a marsupial, Macropus eugenii (Tammar wallaby). J Interferon Cytokine Res 1999, 19:1099–1102.View ArticlePubMedGoogle Scholar
- Harrison GA, Deane EM: cDNA cloning of lymphotoxin alpha (LT-alpha) from a marsupial, Macropus eugenii. DNA Seq 2000, 10:399–403.PubMedGoogle Scholar
- Wedlock DN, Goh LP, Parlane NA, Buddle BM: Molecular cloning and physiological effects of brushtail possum interleukin-1beta. Vet Immunol Immunopathol 1999, 67:359–372.View ArticlePubMedGoogle Scholar
- Hawken RJ, Maccarone P, Toder R, Marshall Graves JA, Maddox JF: Isolation and characterization of marsupial IL5 genes. Immunogenetics 1999, 49:942–948.View ArticlePubMedGoogle Scholar
- Wedlock DN, Aldwell FE, Buddle BM: Nucleotide sequence of a marsupial interleukin-10 cDNA from the Australian brushtail possum (Trichosurus vulpecula). DNA Seq 1998, 9:239–244.PubMedGoogle Scholar
- Cui S, Selwood L: cDNA cloning, characterization, expression and recombinant protein production of leukemia inhibitory factor (LIF) from the marsupial, the brushtail possum (Trichosurus vulpecula). Gene 2000, 243:167–178.View ArticlePubMedGoogle Scholar
- Harrison GA, McNicol KA, Deane EM: Interferon alpha/beta genes from a marsupial, Macropus eugenii. Dev Comp Immunol 2004, 28:927–940.View ArticlePubMedGoogle Scholar
- Zelus D, Robinson-Rechavi M, Delacre M, Auriault C, Laudet V: Fast evolution of interleukin-2 in mammals and positive selection in ruminants. J Mol Evol 2000, 51:234–244.PubMedGoogle Scholar
- Stone WH, Bruun DA, Manis GS, Holste SB, Hoffman ES, Spong KD, Walunas T: The Immunobiology of the Marsupial. Modulators of Immune Responses - The Evolutionary Trail (Edited by: Stolen JS, Fletcher TC, Bayne CJ, Secombes CJ, Zelikoff JT, Twerdok LE and Anderson DP). New Jersey, SOS Publications 1996, 149–165.Google Scholar
- Rowlands DT Jr.: The immune response of adult opossums (Didelphis virginiana) to the bacteriophage f2. Immunology 1970, 18:149–155.PubMedGoogle Scholar
- Stone WH, Brunn DA, Foster EB, Manis GS, Hoffman ES, Saphire DG, VandeBerg JL, Infante AJ: Absence of a significant mixed lymphocyte reaction in a marsupial (Monodelphis domestica). Lab Anim Sci 1998, 48:184–189.PubMedGoogle Scholar
- Beck G: Macrokines:invertebrate cytokine-like molecules? Front Biosci 1998, 3:d559–69.PubMedGoogle Scholar
- Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, Cutts T, Down T, Durbin R, Fernandez-Suarez XM, Flicek P, Graf S, Hammond M, Herrero J, Howe K, Iyer V, Jekosch K, Kahari A, Kasprzyk A, Keefe D, Kokocinski F, Kulesha E, London D, Longden I, Melsopp C, Meidl P, Overduin B, Parker A, Proctor G, Prlic A, Rae M, Rios D, Redmond S, Schuster M, Sealy I, Searle S, Severin J, Slater G, Smedley D, Smith J, Stabenau A, Stalker J, Trevanion S, Ureta-Vidal A, Vogel J, White S, Woodwark C, Hubbard TJ: Ensembl 2006. Nucleic Acids Res 2006, 34:D556–61.View ArticlePubMedGoogle Scholar
- Birney E, Clamp M, Durbin R: GeneWise and Genomewise. Genome Research 2004, 14:988–995.View ArticlePubMedGoogle Scholar
- Burge C, Karlin S: Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997, 268:78–94.View ArticlePubMedGoogle Scholar
- Gross SS, Brent MR: Using multiple alignments to improve gene prediction. J Comput Biol 2006, 13:379–393.View ArticlePubMedGoogle Scholar
- HMMER (http://hmmer.janelia.org) Google Scholar
- Gelfand MS, Mironov AA, Pevzner PA: Gene recognition via spliced sequence alignment. Proc Natl Acad Sci U S A 1996, 93:9061–9066.View ArticlePubMedGoogle Scholar
- Krogh A: Using database matches with for HMMGene for automated gene detection in Drosophila. Genome Res 2000, 10:523–528.View ArticlePubMedGoogle Scholar
- Yeh RF, Lim LP, Burge CB: Computational inference of homologous gene structures in the human genome. Genome Res 2001, 11:803–816.View ArticlePubMedGoogle Scholar
- Stanke M, Schoffmann O, Morgenstern B, Waack S: Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 2006, 7:62.View ArticlePubMedGoogle Scholar
- Korf I, Flicek P, Duan D, Brent MR: Integrating genomic homology into gene structure prediction. Bioinformatics 2001, 17 Suppl 1:S140–8.PubMedGoogle Scholar
- Alexandersson M, Cawley S, Pachter L: SLAM: cross-species gene finding and alignment with a generalized pair hidden Markov model. Genome Res 2003, 13:496–502.View ArticlePubMedGoogle Scholar
- Opossum cytokine sequences (http://bioinf.wehi.edu.au/opossum) Google Scholar
- Gaffen SL, Liu KD: Overview of interleukin-2 function, production and clinical applications. Cytokine 2004, 28:109–123.View ArticlePubMedGoogle Scholar
- Eckenberg R, Rose T, Moreau JL, Weil R, Gesbert F, Dubois S, Tello D, Bossus M, Gras H, Tartar A, Bertoglio J, Chouaib S, Goldberg M, Jacques Y, Alzari PM, Theze J: The first alpha helix of interleukin (IL)-2 folds as a homotetramer, acts as an agonist of the IL-2 receptor beta chain, and induces lymphokine-activated killer cells. J Exp Med 2000, 191:529–540.View ArticlePubMedGoogle Scholar
- Zurawski SM, Vega F Jr., Doyle EL, Huyghe B, Flaherty K, McKay DB, Zurawski G: Definition and spatial location of mouse interleukin-2 residues that interact with its heterotrimeric receptor. Embo J 1993, 12:5113–5119.PubMedGoogle Scholar
- Kruse N, Lehrnbecher T, Sebald W: Site-directed mutagenesis reveals the importance of disulfide bridges and aromatic residues for structure and proliferative activity of human interleukin-4. FEBS Lett 1991, 286:58–60.View ArticlePubMedGoogle Scholar
- Bird S, Zou J, Savan R, Kono T, Sakai M, Woo J, Secombes C: Characterisation and expression analysis of an interleukin 6 homologue in the Japanese pufferfish, Fugu rubripes. Dev Comp Immunol 2005, 29:775–789.View ArticlePubMedGoogle Scholar
- Kaiser P, Poh TY, Rothwell L, Avery S, Balu S, Pathania US, Hughes S, Goodchild M, Morrell S, Watson M, Bumstead N, Kaufman J, Young JR: A genomic analysis of chicken cytokines and chemokines. J Interferon Cytokine Res 2005, 25:467–484.View ArticlePubMedGoogle Scholar
- Farrar JD, Asnagli H, Murphy KM: T helper subset development: roles of instruction, selection, and transcription. J Clin Invest 2002, 109:431–435.PubMedGoogle Scholar
- Buddle BM, Young LJ: Immunobiology of mycobacterial infections in marsupials. Dev Comp Immunol 2000, 24:517–529.View ArticlePubMedGoogle Scholar
- Jansen AM, Madeira FB, Deane MP: Trypanosoma cruzi infection in the opossum Didelphis marsupialis: absence of neonatal transmission and protection by maternal antibodies in experimental infections. Mem Inst Oswaldo Cruz 1994, 89:41–45.View ArticlePubMedGoogle Scholar
- Gomes DC, da Cruz RP, Vicente JJ, Pinto RM: Nematode parasites of marsupials and small rodents from the Brazilian Atlantic Forest in the State of Rio de Janeiro, Brazil. Rev Bras Zool 2003, 20:699–707.View ArticleGoogle Scholar
- Beveridge I, Arundel JH: Helminth Parasites of Grey Kangaroos, Macropus Giganteus Shaw and M. Fuliginosus (Desmarest), in Eastern Australia. Aust Wildlife Res 1979, 6:69–77.View ArticleGoogle Scholar
- Vu MD, Li XC: The common gamma chain-cytokines and transplantation tolerance. Arch Immunol Ther Exp (Warsz) 2004, 52:267–273.Google Scholar
- Wang Z, Vandeberg JL: Immunotolerance in the laboratory opossum (Monodelphis domestica) to xenografted mouse melanoma. Contemp Top Lab Anim Sci 2005, 44:39–42.PubMedGoogle Scholar
- Lebedeva IV, Su ZZ, Chang Y, Kitada S, Reed JC, Fisher PB: The cancer growth suppressing gene mda-7 induces apoptosis selectively in human melanoma cells. Oncogene 2002, 21:708–718.View ArticlePubMedGoogle Scholar
- Pearse AM, Swift K: Allograft theory: transmission of devil facial-tumour disease. Nature 2006, 439:549.View ArticlePubMedGoogle Scholar
- Broad Institute - Opossum genome (http://www.broad.mit.edu/mammals/opossum/) Google Scholar
- Henikoff S, Henikoff JG: Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 1992, 89:10915–10919.View ArticlePubMedGoogle Scholar
- Pearson WR, Lipman DJ: Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A 1988, 85:2444–2448.View ArticlePubMedGoogle Scholar
- Eddy SR: Profile hidden Markov models. Bioinformatics 1998, 14:755–763.View ArticlePubMedGoogle Scholar
- Krogh A, Mitchison G: Maximum entropy weighting of aligned sequences of proteins or DNA. Proc Int Conf Intell Syst Mol Biol 1995, 3:215–221.PubMedGoogle Scholar
- Felsenstein J: PHYLIP-Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5:164–166.Google Scholar
- NetOGlyc (http://www.cbs.dtu.dk/services/NetOGlyc) Google Scholar
- NetNGlyc (http://www.cbs.dtu.dk/services/NetNGlyc) Google Scholar
- PROSITE database (http://au.expasy.org/prosite/) Google Scholar
- Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22:4673–4680.View ArticlePubMedGoogle Scholar
- Jones DT, Taylor WR, Thornton JM: The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 1992, 8:275–282.PubMedGoogle Scholar
- Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 2004, 5:150–163.View ArticlePubMedGoogle Scholar
- Needleman SB, Wunsch CD: A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 1970, 48:443–453.View ArticlePubMedGoogle Scholar
- Nicholas KB, Nicholas HBJ: GeneDoc: a tool for editing and annotating multiple sequence alignments. 1997., http://www.psc.edu/biomed/genedoc: Google Scholar
- ANGIS (http://www.angis.org.au) Google Scholar
- Bazan JF: Unraveling the structure of IL-2. Science 1992, 257:410–413.View ArticlePubMedGoogle Scholar
- Walter MR, Cook WJ, Zhao BG, Cameron RP Jr., Ealick SE, Walter RL Jr., Reichert P, Nagabhushan TL, Trotta PP, Bugg CE: Crystal structure of recombinant human interleukin-4. J Biol Chem 1992, 267:20371–20376.PubMedGoogle Scholar
- Eisenmesser EZ, Horita DA, Altieri AS, Byrd RA: Solution structure of interleukin-13 and insights into receptor engagement. J Mol Biol 2001, 310:231–241.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.